目前整套下來已知:
- 前備知識:NGS+次世代定序相關原理與知識(web lab 部份沒有講很多)
當我們理解了fq數據之後,做這些過濾就不會很難,你也完全可以自己編寫工具來進行個性化的過濾。目前也已有很多工具用來Adapter和低質量鹼基,比如Trimmomatic、SOAPnuke、cutadapt、untrimmed等不下十個,今天想介紹一下我有用過的Fastq。
PS.大部份工具原理:就是通過滑動一定長度的窗口,計算窗口內的鹼基平均質量,如果過低,就直接往後全部切除,注意,不是挖掉read中的這部分低質量序列,而是像切菜一樣,直接從低質量區域開始把這條read後面的所有其它鹼基全!部!切!掉!!!!!
否則就是在人為改變實際的基因組序列情況。
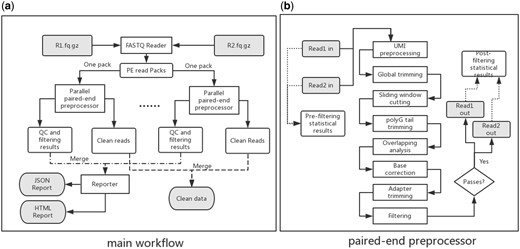
首先:前備概念要知道定序出來的FASTQ檔,需要品質控制和預處理,保證接下來,下游分析輸入數據都是乾淨可靠的。
Fastp = FASTQC(質控)+cutadapt(去除接頭)+Trimmomatic(剪裁)+預處理....
Fastp是基於C++,支持多線程,包含fastQC和Trimmomatic的一些功能。
為多線程並行處理而設計的,每個線程都有一個單獨的環境來存儲它處理的讀取的統計值。在處理完所有操作後,這些值將被合併,報告器將生成HTML和JSON格式的報告。Fastp報告預過濾和後過濾數據的統計值,以便於比較過濾完成後數據質量的變化。
Fastp支持SE (single-end)和PE (paired-end)數據。
fastp -i in.fq -o out.fq
fastp -i in.R1.fq.gz -I in.R2.fq.gz -o out.R1.fq.gz -O out.R2.fq.gz
usage: fastp -i <in1> -o <out1> [-I <in1> -O <out2>] [options...]
options:
# I/O options
-i, --in1 read1 input file name (string)
-o, --out1 read1 output file name (string [=])
-I, --in2 read2 input file name (string [=])
-O, --out2 read2 output file name (string [=])
PS. 更詳細使用可以到github文檔裏面看
自動生成質控文件fastp.html和fastp.json,可以通过--html 和--json指定文件名字。
thread 默認為2,可以設定 -w, --thread ,最大為16
fastp -i in.R1.fq.gz -I in.R2.fq.gz \
-o out.R1.fq.gz -O out.R2.fq.gz \
--umi \
--umi_loc per_read \
--umi_len 6 \
--umi_prefix UMI \
--umi_skip 4
--umi # enable umi
--umi_loc per_read # read1和read2都有UMI
--umi_len 6 # UMI序列長度
--umi_prefix UMI #UMI序列前綴
--umi_skip 4 # UMI序列後要切除的鹼基數


 iThome鐵人賽
iThome鐵人賽