最新版本請見:https://chenhsieh.com/post/bioinfo/07-sra-2/
為了讓農民更開心、健康地生產香蕉,挑戰者要找到相關的文獻,了解香蕉被有益微生物感染之後,會產生怎樣的基因調控。其中用以推估基因表現量的高通量定序方式,RNA-seq,的原始資料都會公開在傳說中的 SRA 大秘寶平台,想要取得寶藏就必須使用專屬的 SRA Toolkit 來下載,究竟能否成功呢?
SRA 是 Sequence Read Archive 的縮寫,這個資料庫上存放生物序列資料並且開放所有研究社群存取,期能提高試驗的可再現性,及提供一個跨研究資料比較的可能性。SRA 存放高通量定序的原始定序資料及比對 (alignment) 結果資料,目前已經包括 Roche 454 GS System®, Illumina Genome Analyzer®, Applied Biosystems SOLiD System®, Helicos Heliscope®, Complete Genomics®, and Pacific Biosciences SMRT® 等等。
SRA 隸屬於 International Nucleotide Sequence Database Collaboration (INSDC)。每天接收、處理、封存數千筆定序資料,以 terabytes 為計數單位。上述資料來源包括 NCBI Sequence Read Archive (SRA), European Bioinformatics Institute (EBI), 還有 DNA Database of Japan (DDBJ),三大平台的資料都是共享的。
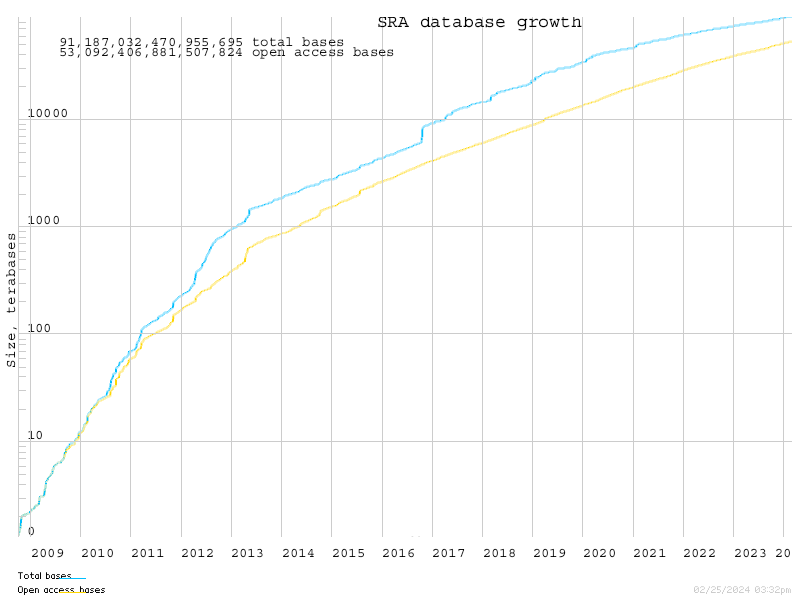
(SRA 資料庫封存資料量之成長,圖片來源: SRA database growth)
在下方連結之頁面,選取相對應作業系統下載 SRA Toolkit,以及最下方的 md5 checksums
https://www.ncbi.nlm.nih.gov/sra/docs/toolkitsoft/

校驗碼 (checksums) 是依據檔案內容所產生的一組編碼,檔案內容有些為更動的話校驗碼就會完全不同,因此用以前後比對確保檔案傳輸結果。SRA Toolkit 下載完成,開啟終端機,cd Download使用 md5 指令來計算該檔案的校驗碼,md5 sratoolkit.current-mac64.tar.gz,檢查回傳的結果與網站上公布的是否一樣。小技巧:輸入終端機指令的時候,如果檔案名稱太長,可以輸入了開頭之後,按 tab 來讓終端機幫你補上可能的檔案名稱參數。
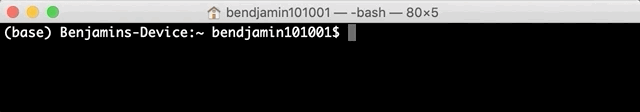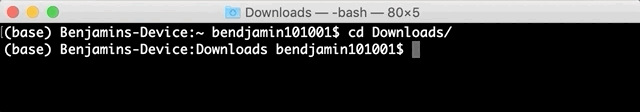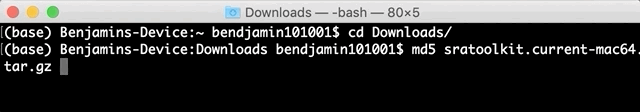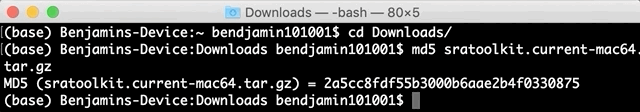
看來結果與網站上顯示的 mac64 檔案之校驗碼一樣。
01f66e1a7867b7efc09def4b7484b36d *sratoolkit.current-centos_linux64.tar.gz
2a5cc8fdf55b3000b6aae2b4f0330875 *sratoolkit.current-mac64.tar.gz
ba91fdcebe84e48e9a42fcdf0807e032 *sratoolkit.current.revision
07a6eea17ad126c1ce4c92718ffe8600 *sratoolkit.current-ubuntu64.tar.gz
55dc5dcffdf13691967a830ba3742990 *sratoolkit.current.version
57f41130498dbdcd8a4cec52ab7dda88 *sratoolkit.current-win64.zip
安裝與基本設定說明網頁
https://trace.ncbi.nlm.nih.gov/Traces/sra/sra.cgi?view=toolkit_doc&f=std
解壓縮之後,cd 進入解壓縮的資料夾之 bin,或是將 bin 的路徑加入到系統環境變數,就可以直接呼叫其中的指令。
首先來到 BMC genomics 的期刊首頁,搜尋 banana, rhizobacteria 出來的第一筆結果就是神秘文章

在瀏覽器上直接搜尋關鍵字 availability,共三筆結果,前兩筆在內文中,顯然不是我們要的
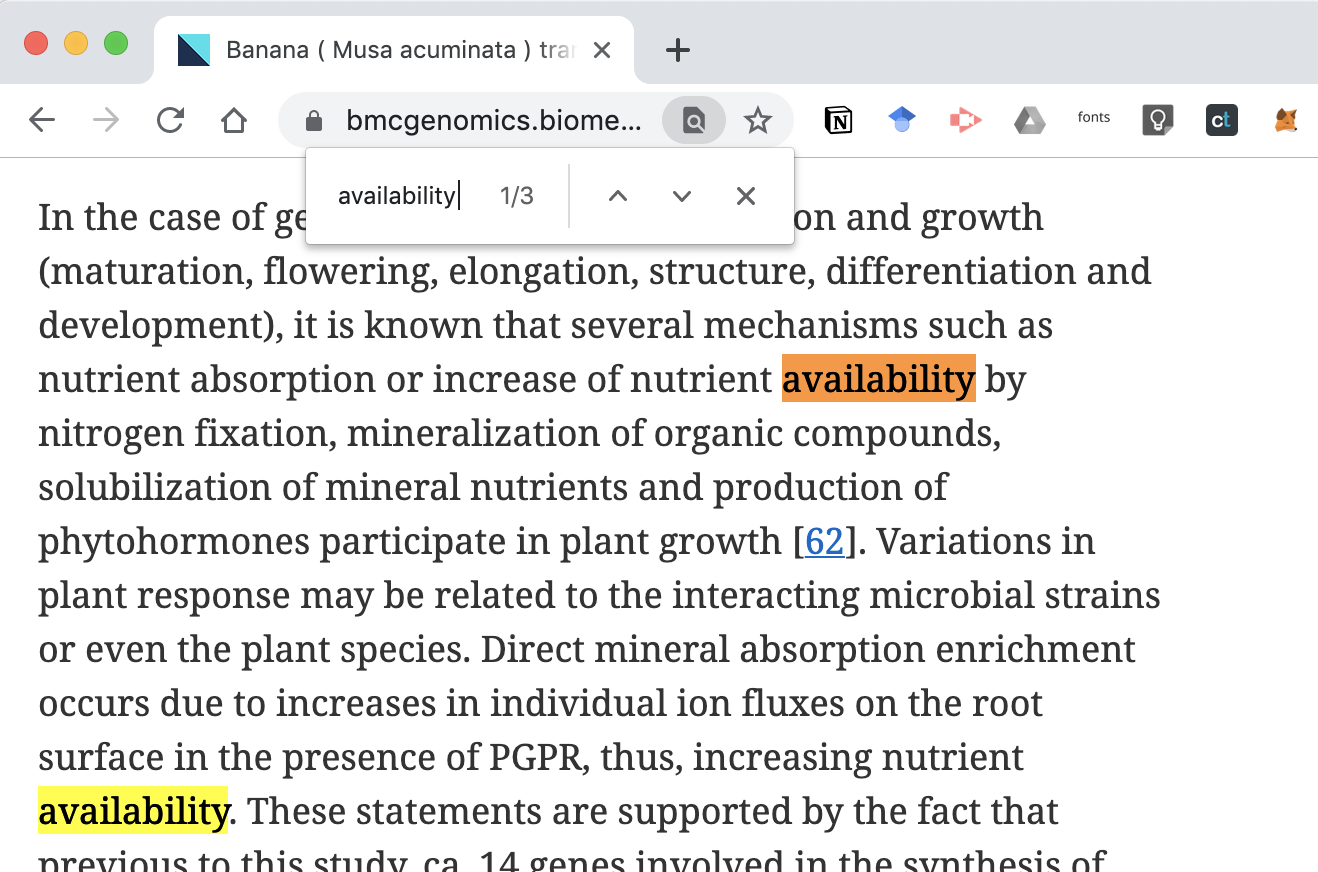
第三筆結果 Availability of data and materials,第一句內文就提供了 Gene Express Omnibus 的連結
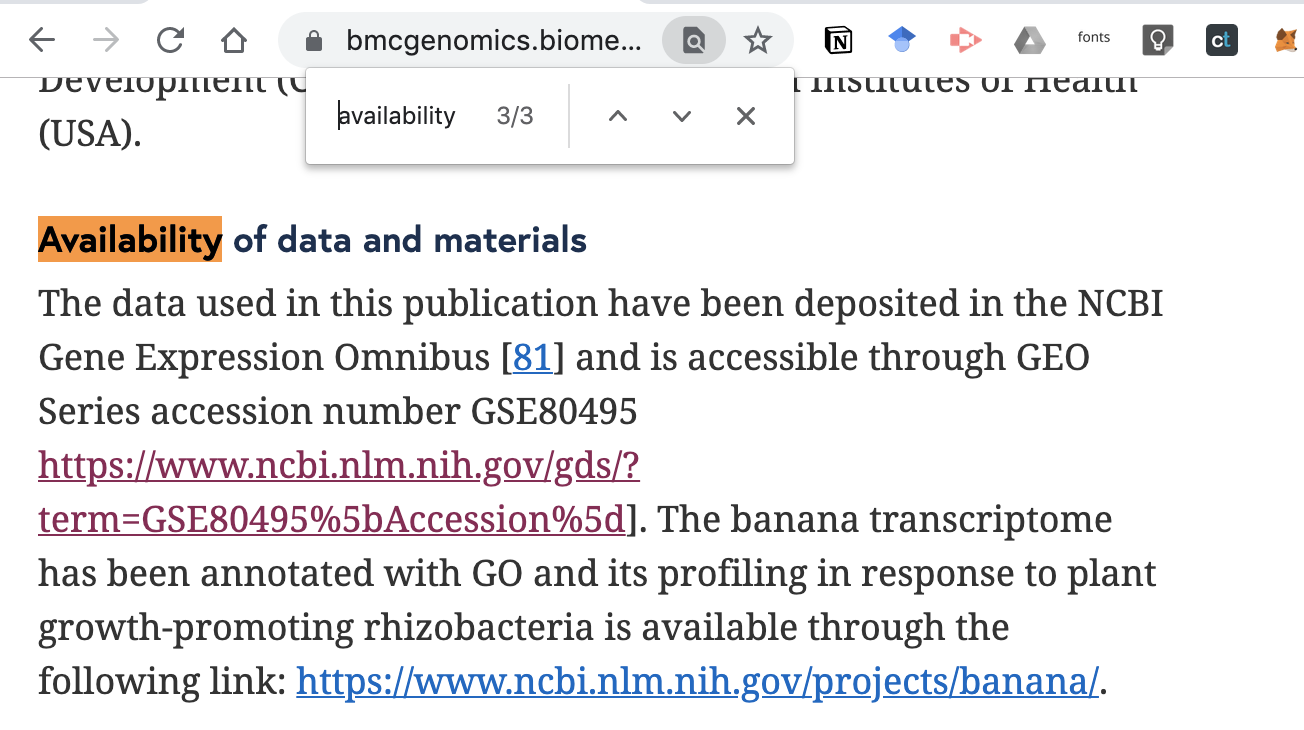
於連結中以瀏覽器搜尋 SRA,點下 SRA Run Selector
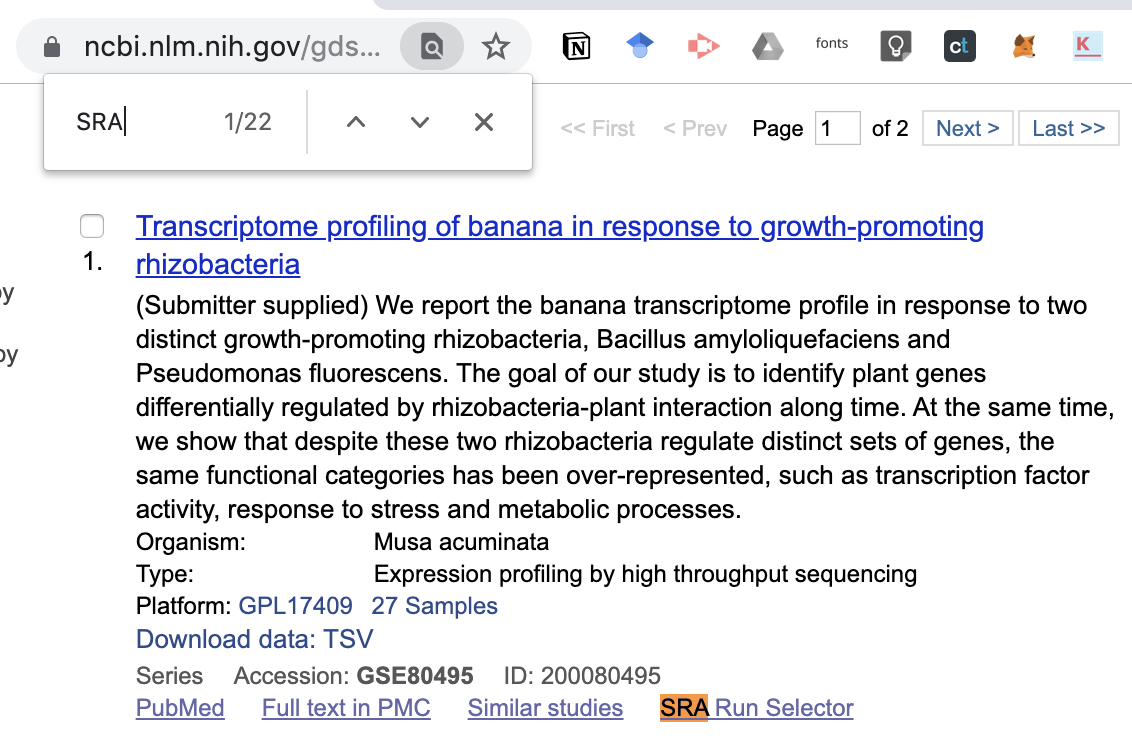
來到 NCBI 已經持續改版到相當友善的資訊呈現頁面
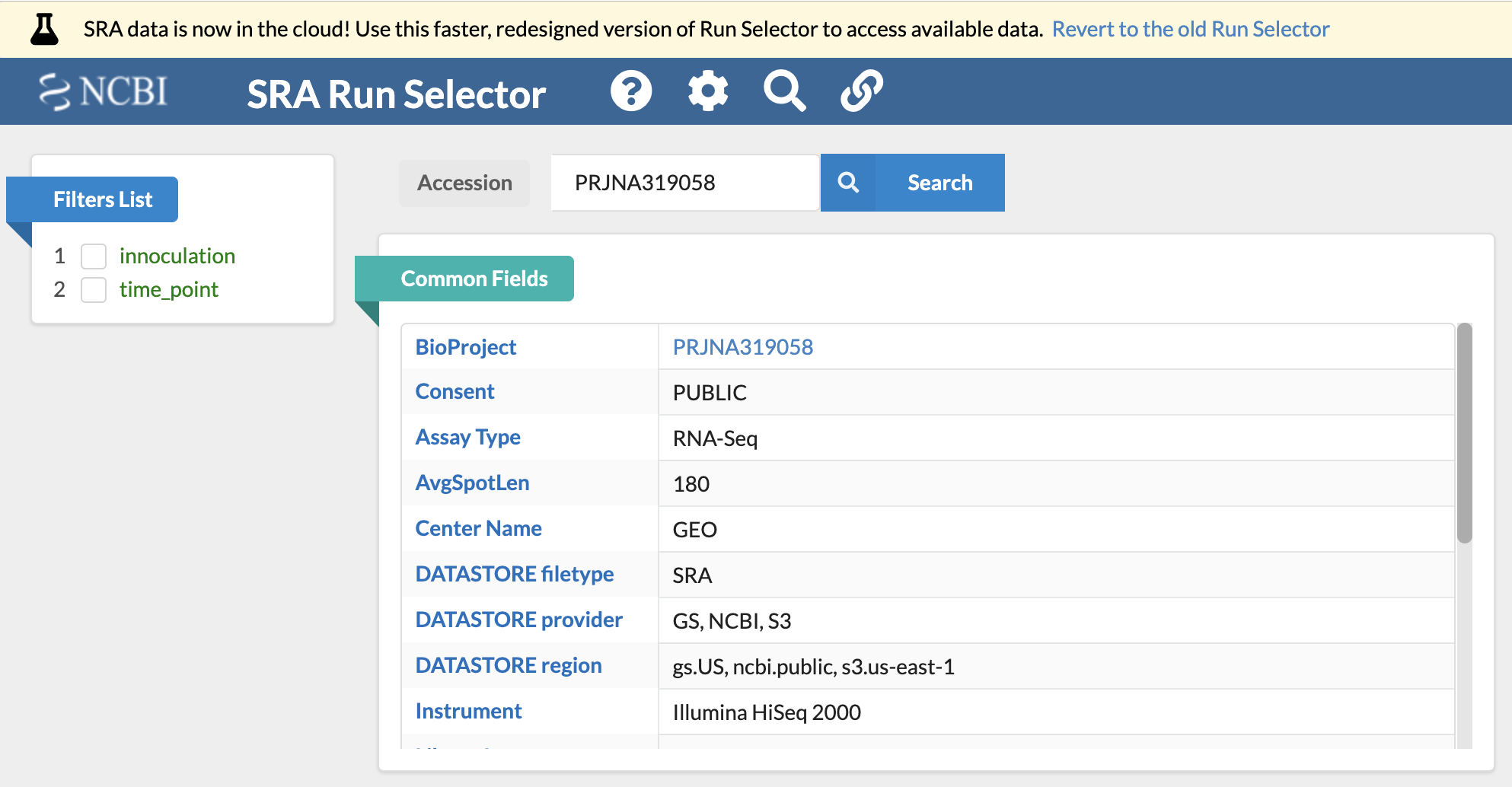
下方有一個大表格說明各個 Run 的資訊,先記下第一個 SRR3406492 吧
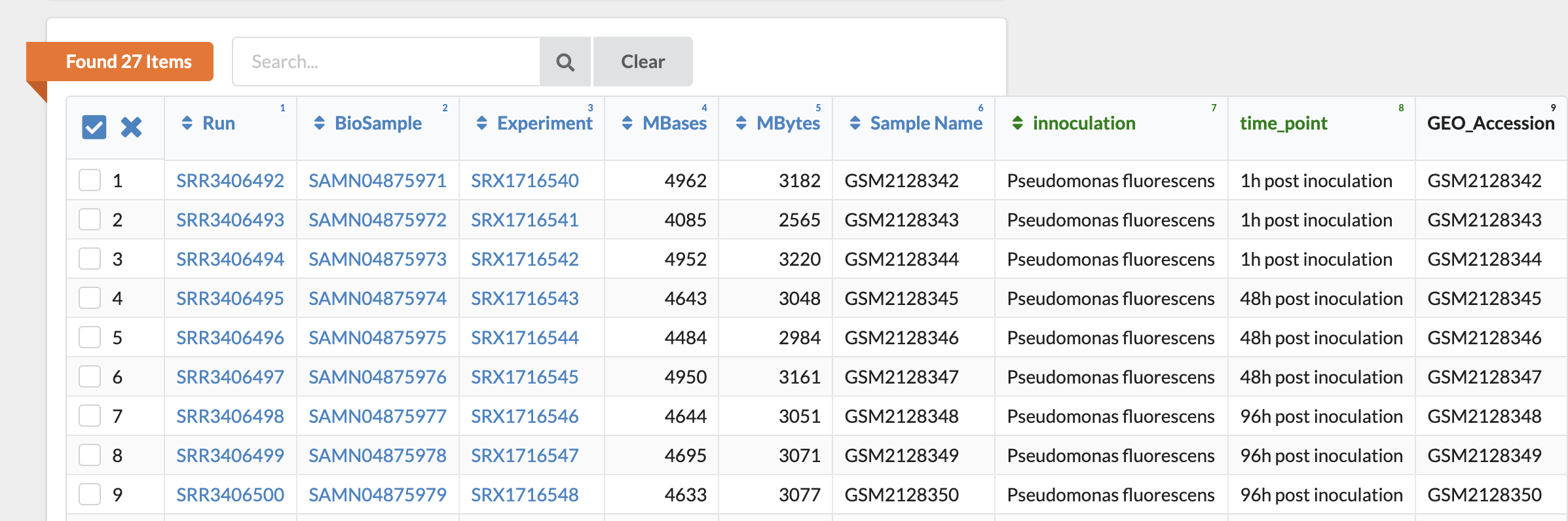
我們這裡先偷吃步一下,不要下載完整的 SRR3406492,因為他有 4.96 G 個序列文字,檔案大小則是 3.11 Gb。使用 -X 參數,後方加上數字,就可以只先下載該 Run 中的頭幾筆資料。—split-files 則是將 paired-end 次世代定序的左端以及右端分開儲存於不同的檔案中,這對後續分析也比較方便。
fastq-dump -X 5 --split-files SRR3406492
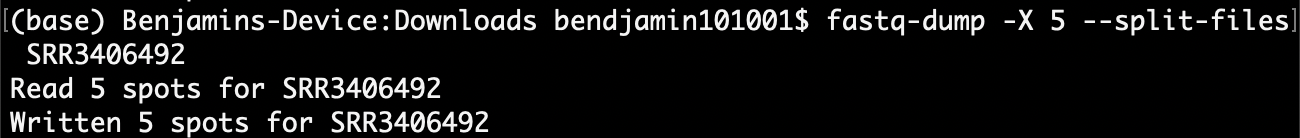
終端機回傳 Written 5 spots for SRR3406492 代表執行成功並且輸出成檔案。
直接用右鍵點選 fastq 檔案,用 Atom 開啟吧!
至於這些亂碼般的文字代表什麼,我們將於下一回「轉錄體流程篇」的第一回中說明~
Studies : Browse : Sequence Read Archive : NCBI/NLM/NIH
Toolkit Documentation : Software : Sequence Read Archive : NCBI/NLM/NIH
Linux 產生 MD5 與 SHA1 校驗碼 Checksum 使用教學,檢查檔案是否損毀 - G. T. Wang
Download SRA sequences from Entrez search results
關於作者
謝晨 (Chen Hsieh),臺大園藝暨景觀學系研究所碩士。讀碩士前的興趣是懷著寫點程式妄圖解決農業問題的夢想參加比賽,拿了幾個黑客松與 Open Data 創新應用競賽的獎,卻都沒有勇氣將項目經營下去;研究所期間的興趣轉換成讀學術期刊的出刊電子報。靠著這些興趣當選 107 學年的臺大優秀青年,畢業後在農場旁的研究館辦公室寫點東西,希望可以跟世界分享生物資訊與園藝的樂趣!
感謝選擇匿名的朋友協助校閱初稿與提供意見,也敬請各位讀者不吝指教!
• Website: ChenHsieh.com


 iThome鐵人賽
iThome鐵人賽