題目連結:https://rosalind.info/problems/grph/
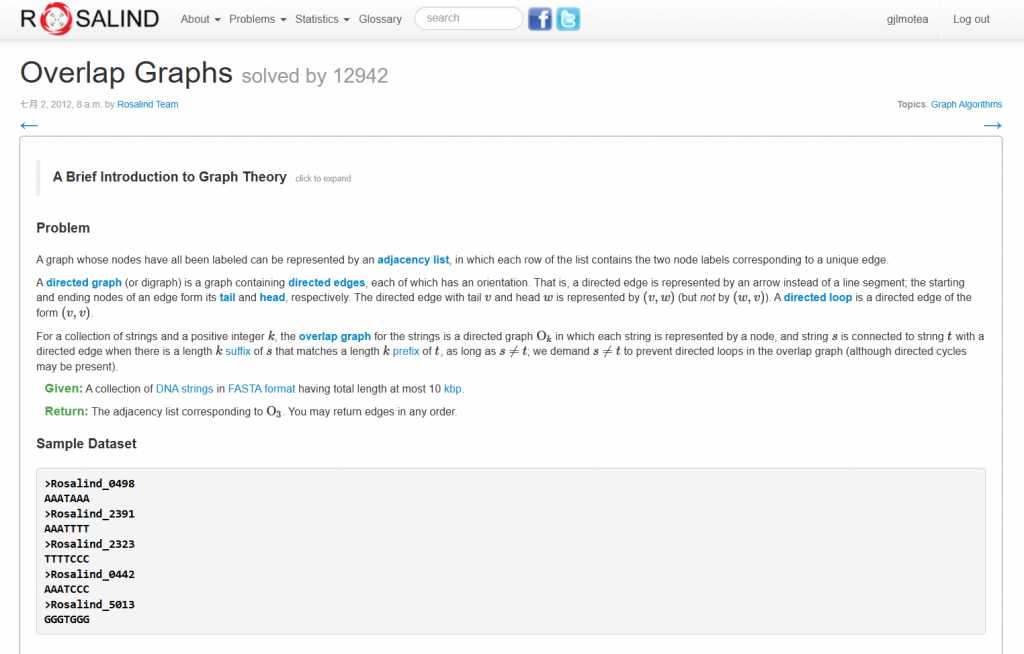
直接來看輸入與輸出
輸入
>Rosalind_0498
AAATAAA
>Rosalind_2391
AAATTTT
>Rosalind_2323
TTTTCCC
>Rosalind_0442
AAATCCC
>Rosalind_5013
GGGTGGG
輸出
Rosalind_0498 Rosalind_2391
Rosalind_0498 Rosalind_0442
Rosalind_2391 Rosalind_2323
這一題是重疊圖,其中提到O3,等於overlop 3個元素(長度k=3)
就是指,「1號序列的前三碼,若與2號序列的末三碼一樣」,則將其標示出來
可返回任意排序內容
直觀一點的話,想像有鋸齒狀卡住的結構,一股力量往左扯、下面一股力往右扯
上面有三個元素、下面也三個元素
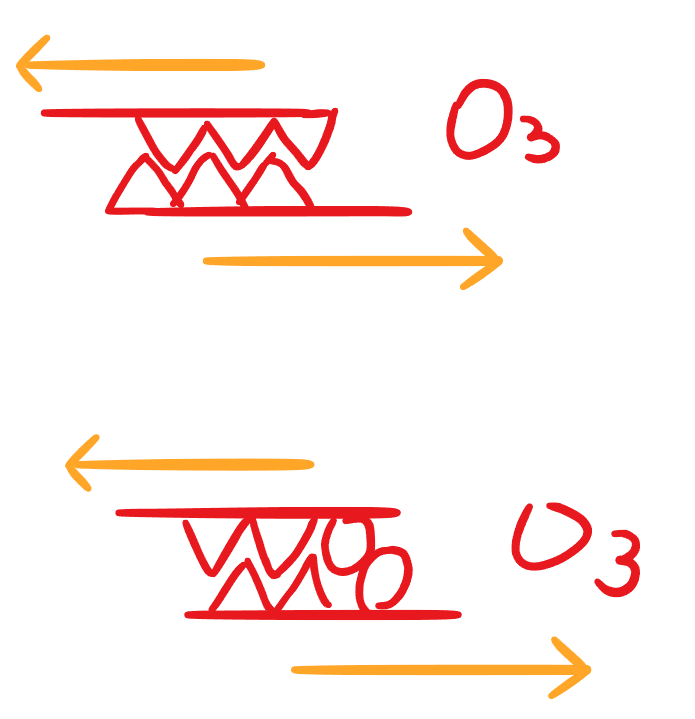
O3關係就是有三個元素,上下互相卡的很緊
卡越緊代表重疊度很高、很overlap(不是"很over",是overlap)
那麼再看這張圖,上下元素數量來到四個
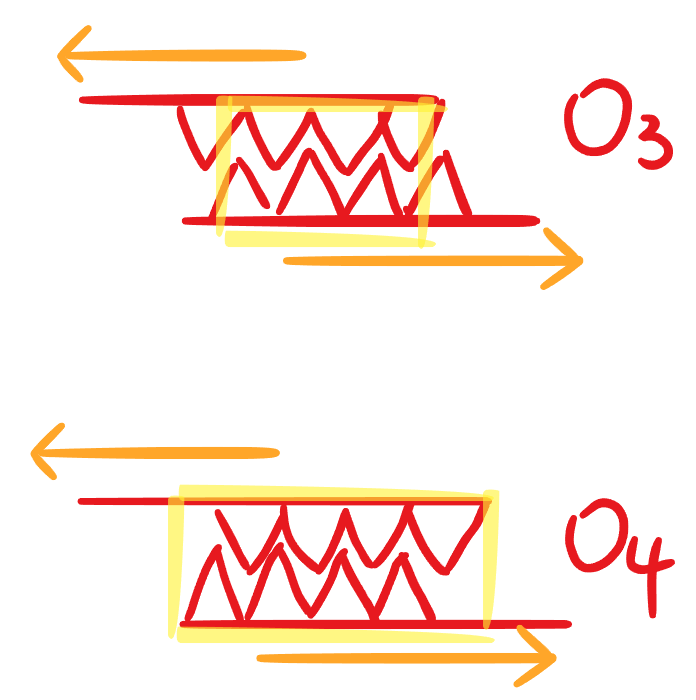
可以形成O3關係、也可以形成O4關係
O4的話卡死更緊了呢,代表更overlap了
結論:AAACTTT與CTTTCCC => 不是O3關係,而是O4關係,因為重複了CTTT
AAATTTT與TTTTCCC => 是O4關係,重複了TTTT
但他們同時也可以形成O3關係TTT、O2關係TT、O1關係T
在圖論或組裝實作中通常只記「最大重疊」,以避免歧義與重複邊。但在這一題中只考慮O3邊,所以也需將O3關係列入計算
但是要小心
適用於O4(長)關係的,則不一定適用於O3(短)關係
反之,不適用於O3(短)關係的,不代表一定無法形成O4(長)關係
要看兩者的重複結構關係
O(n²) 的解法,直接把A與B兩兩配對,將每一個組合全部列出比對
符合O3關係的就抓出來!
程式碼:
data = """
>Rosalind_0498
AAATAAA
>Rosalind_2391
AAATTTT
>Rosalind_2323
TTTTCCC
>Rosalind_0442
AAATCCC
>Rosalind_5013
GGGTGGG
"""
def parse_fasta(data: str) -> dict:
sequences = {}
label = ""
for line in data.strip().splitlines():
line = line.strip() # 縮排防呆
if not line: # 跳過空行
continue
if line.startswith(">"):
label = line[1:].strip() # 去除label頭尾空白
if not label: # 跳過空標籤(不合法)
label = ""
continue
sequences[label] = ""
elif label:
sequences[label] += line
return sequences
seqs = parse_fasta(data)
for s1 in seqs:
for s2 in seqs:
if s1 == s2:
continue
if seqs[s1][-3:] == seqs[s2][:3]:
print(s1, s2)
這一題雖然是在建 Overlap Graph 重疊圖運算,只是在計算前後是否相等,但其實更像在做「拼圖的組裝」,將每一塊拼圖以合理的方式拼建起來。

類似次世代定序(NGS)將多個小片段,依照最有可能的組合方式拼起來,將拼每一個Reads(NGS產生的短片段序列)拼裝出原本一整條基因的樣貌
次世代定序 (NGS) 的組裝流程:
因為技術上,仍無法一次直接讀完整條基因體
而且目前技術讀的時候,無法做到100%精確
第二代定序(NGS)短讀長,市占率最高
第三代定序(TGS)長讀長,至今仍然在發展,尚未完全成熟和普及
坦白說,所以我們這一題O₃ overlap graph 每個read兩兩比對的解法
只適合小資料驗證概念,因為用在NGS的話,運算量會非常可觀(幾百萬甚至幾億條reads),運算時間非常嚇人
次世代定序 (NGS, Next-Generation Sequencing):
一次只能讀大約數百個以下的bp
Illumina公司的SBS(Sequencing by Synthesis):準確度極高,但讀長短,需大量拼接。讀長50–200bp,再繼續往下讀精確度大幅降低(訊號誤差會放大累積)
SOLiD (Applied Biosystems / Life Technologies / 被Thermo Fisher收購):ligation探針,讀長35–75bp,已被市場淘汰
Ion Torrent (被Thermo Fisher收購):半導體晶片定序,偵測H⁺釋放,讀長100–400bp,已被市場淘汰
第三代定序 (TGS, Third-Generation Sequencing):
一次能讀非常長的bp,但仍需克服精準度問題
PacBio公司的SMRT:利用零模波導 (Zero-Mode Waveguide, ZMW),觀測單一聚合酶合成DNA過程,讀長可達10000-100000bp,HiFi Reads大幅提升準確度
Oxford Nanopore公司的Nanopore:讓DNA分子通過奈米孔,偵測電流阻抗變化以辨識核苷酸,讀長可達數十萬甚至幾乎無上限
基因體長度(genome):
人類基因體大約30億個鹼基對(bps),DNA拉直大約能達1公尺長
所以做基因定序時,會將同一份基因體(Genome)拷貝許多次之後、打碎再讀取
之後再盡可能讀取
目前定序魔王在於:
同聚物序列(AAAAA…)難以判讀,因為電流訊號太相似,難以準確判斷「到底是5個A,還是7個A」,而目前市面上存活的這些技術都能部分克服、或者繞過這個難題,以達到精準
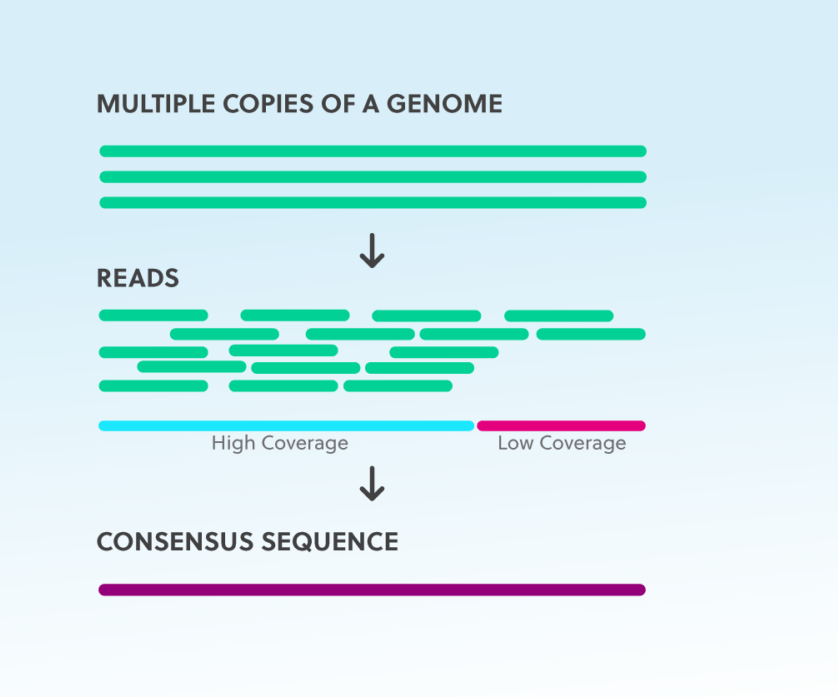
看圖可以發現,我們無法完全把每個copy(複本)上的所有序列都讀過一遍,因為打碎之後還要蒐集全部碎片是不務實的
只能做到盡量cover,反正只要copy的數量充足,定序時讀取足夠多次
以概率來說,就能夠cover整條genome
所以在實驗設計時會要求coverage數值,以降低誤差
30X coverage => 每個鹼基平均會被讀到30次
100X coverage => 每個鹼基平均會被讀到100次
即使個別read有可能錯,只要覆蓋率夠高就能夠平均、抵銷掉誤差
單位到底是用「bps還是nts」
bp (base pair):雙股 DNA 的長度單位。
nt (nucleotide):單股核酸(單股DNA或RNA)的長度單位
兩者都是在指序列的長度
嚴格來說,1bp = 2nts,因為一個「鹼基配對」是由「2個核甘酸互補」組成
由於至今所有的定序方式都是讀取「單股」辨識,沒辦法讀雙股黏在一起的序列
就有點像,讀書總要把書本攤開、分離成左右兩個頁面,才能讀取上面的文字
雖然定序機器讀的都是單股,但要寫nt還是bp取決於樣本最終是RNA還是DNA
RNA定序(讀的是RNA)就寫nt,DNA定序(讀的是雙股DNA)就寫bp
...嚴格來說,理應如此啦!
但是至今不論DNA或RNA,用的「普遍都是bp」
就像服飾店講「一條拉鍊」,沒有人糾正「應該是一對齒鏈」
去服飾店裡問,阿姨您今天修理了幾條拉鏈?
老闆娘馬上就回答你了
通常老闆娘不會反問:「你到底是問幾對拉鍊、還是幾條拉鏈?」但其實一條拉鍊是由兩排齒鏈互相咬合,嚴格來說應該是一對,由兩條鏈帶組成
PS 拉鏈是1917年Gideon Sundbäck的專利