
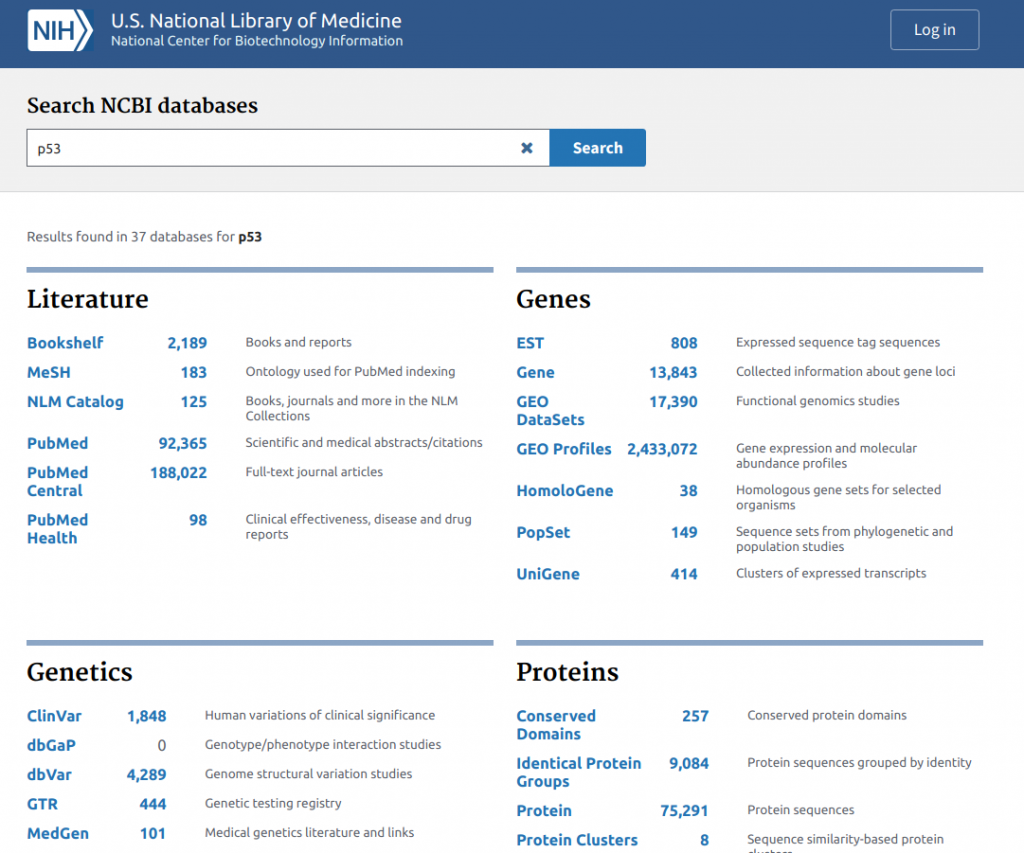
NCBI(National Center of Biotechnology Information的縮寫)— 在搞生物研究的圈子裡,乃是無人不知無人不曉的資料庫。你在上面隨便搜尋一個關鍵字(通常是跟生命科學有關的字詞),它會馬上搜尋它底下所關聯的幾十個資料庫,看有哪些紀錄與你給出的關鍵字有關,從基因、蛋白質、研究文獻到有哪些物種身上可能有這個東西,都會一一列給你,舉例說:我想知道所有一切跟癌症抑制基因—p53有關的所有資訊,你在NCBI上搜尋p53,你會看到這種畫面:
這個問題說實在得問施主您想幹麻?(喂~)
通常我們之所以會用到NCBI找資料的目的挺雜的:有的人想找某個(些)基因的DNA/蛋白質序列,有的人想找某個遺傳變異位點落在什麼基因上或與什麼疾病有關,也有人想知道的是有哪些物種會有他現在想問的這個基因或蛋白質或是類似功能的分子。以我自己日常會運用到NCBI的例子來說,我比較需要會用到的是不同資料庫之間accesession number或是ID之間的對應關係。
我以我前陣子在處理的一種資料中的ID對應及轉換作為一個案例來分享:
自然界裡頭充滿了各種微生物,相信大家是知道的。其中呢,我們最常聽到的一些buzzwords中,什麼益生菌、腸道菌等應該也是各位耳熟能詳的。我們這些在生物領域的人在透過大量的研究之後,發現這些小的不得了的微生物竟然在人體中佔了1~3%的體重,還會以相當大的程度影響到人體的健康程度,在這情況下,我們就會想了解健康的人與不健康的人身上這些原住民的組成有什麼差異?有哪些細菌會讓我們不容易變胖?有哪些菌可能會讓我們特別容易拉肚子?關心這些議題,同時利用次世代定序(Next generationa sequencing)技術加生物資訊分析來幫我們看看我們研究的樣本裡面到底有哪些細菌。在這過程中,我們這些搞生物資訊的人,就會先處理這些定序出來的序列資料,再比對(alignment)回那些資料庫中的細菌基因序列,透過一些標準來篩選最終的結果後,我們大概就可以得知在這個樣本中各種細菌的物種組成比例。不過呢~這個時候也往往因為所選取使用的序列來源資料庫的關係,在做完比對後,我得到的可能是某個資料庫的ID,並不是最終那些細菌所對應的物種分類資料庫的ID。因此,我就得先回NCBI上找這兩種ID之間的對應關係。所幸的是,目前在NCBI上這種資料蒐集得很全面,而且他們還提供了相當友善的API,讓我們能夠透過這個API查詢、爬取我們所需的資料。不僅如此,他們還提供了他們自己開發的指令工具—E-utilities讓我們只要下command就可以拿到我們需要的資料啦!
在BioJulia這個偉大的專案中,有個叫做BioServices.jl的package,採用了HTTP、XMLDict跟JSON這幾個packages加上NCBI提供的API實做出E-utilities的功能。以下我們就來看看在Julia底下怎麼使用吧!
BioServices.jl
using Pkg
Pkg.add("BioServices.jl")
einfo()抓取PubMed裡頭所有的資料,並將結果輸出到pubmed.xml這個檔案中:res = einfo(db="pubmed")
write("pubmed.xml", res.data)
efetch()抓取NM_001126.3這個基因的資料,同時使用EzXML去parse返回的XML,最終抓取DNA序列:res = efetch(db="nuccore", id="NM_001126.3", retmode="xml")
using EzXML
doc = parsexml(res.body)
seq = findfirst("/GBSet/GBSeq", doc)
using BioSequences
DNASequence(content(findfirst("GBSeq_sequence", seq)))
這點我在鐵人挑戰賽剛開始的時候就講過了,BioJulia這個專案底下的很多Packages說明文件都有錯誤,以至於當你照著文件來搞確只能得到錯誤訊息,我也是邊學邊用邊修改他的文件,所以在這之前,也請各位在使用前多注意了。今天就先這樣啦~
請問在 NCBI 下載大量資料時有沒有比較好的方式?
有時遇到自行 query 的結果筆數非常大時,常常都需要重新下載好幾次。
是否有推薦的下載方法或是程式?
如果真要大量下載的,直接去NCBI FTP吧~這大概是最安全又可靠的方式了,嫌速度慢,可以搭配aspera來用
原來還有 apera 這個方法,感謝。
ftp 下載基本上是沒什麼問題。而aspera 的機制似乎也只能運用在已存在伺服器上的 data?
之前的解法是自己寫 script分批慢慢下載,必要時中間還得停頓個幾秒...

 iThome鐵人賽
iThome鐵人賽