以往我們在說NGS—也就是所謂的次世代定序時,所使用的樣品DNA其實都是來自某塊組織裡的細胞(假設使用的是人類的樣品),還記得以往我們學生物學時學過,人體是由好幾個系統所構成(呼吸系統、循環系統等),而系統又是由幾個不同的器官所構成,器官則是由不同的組織所構成,那構成這些組織的就是許多種不同的細胞。遺傳學告訴我們,人體中每個不同的細胞都擁有同樣的DNA,那又是怎麼決定哪些細胞要變成神經細胞、哪些細胞要成為血球細胞的呢?欸,這就是生物學比起其他學科相對複雜很多的原因。同樣的,回到原本NGS的話題,由於不同種類的細胞其基因表達的profile也不同,所以過去我們切下了腫瘤組織,要與所謂的正常組織細胞來比較其轉錄體的差異時,當真是一種很粗略的推論。因此,當單細胞定序的技術開始成熟之後,科學家是多麼地興奮,多想趕快把人體中所有的細胞都定序過一遍。但是啊~事情不是我們想的這麼簡單,在一塊組織細胞裡頭,所函有的細胞種類或是亞型的數量可說是超過我們以往的認知,所以單細胞定序的研究當中所面臨的第一個難題就是怎麼區分出不同種類的細胞,好讓我們接下來才可以問:這種細胞與那種細胞的基因表達有什麼差異。而人體中有2萬多個基因,單細胞定序所使用的每塊組織中往往又有上萬顆細胞,然後我們可能得比較兩種不同conditions底下,這些細胞當中我們真正有興趣的那些細胞有何差異,這個問題所引伸出的難題就是龐大的計算量。雖然這兩三年也陸續有許多工具被發表,但共同的問題就是怎麼將這塊的計算效率提昇,所幸,有人使用Julia實作了一個工具來幫我們處理這樣的難題,那就是今天要介紹的CellFishing.jl。
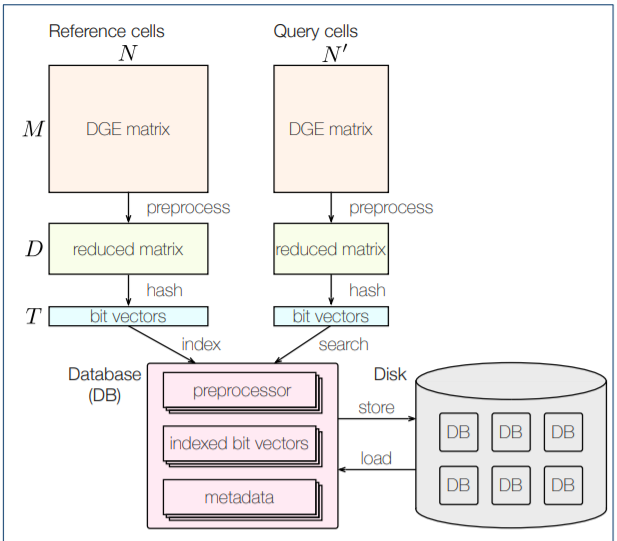
作者們主要的想法就是先將reference cell type的資料庫建立好,隨後只要把query cell的資料拿過來跟資料庫做比對,就可以知道這是哪種細胞啦!概念是簡單的,不過實作起來卻不簡單,首先要先將原始的gene expression matrix用SVD做預處理及降維,再透過locality-sensitive hashing這個方式把降維後的資料存成bit vector,最後存進資料庫中。
CellFishing.jl的使用也很簡單,但必須先將原始的定序資料透過生物資訊方法轉換成表現量數據,再以這些數據作為input丟進CellFishing.jl來處理:
# Import packages.
using CellFishing
using CSV
# Load expression profiles of database cells.
# Note: We highly recommend using the Loom format (http://loompy.org/) to
# load expression data, because loading a large matrix in plain text takes
# extremely long time.
data = CSV.read("database.txt", delim='\t')
cellnames = string.(names(data))
featurenames = string.(data[:,1])
counts = Matrix(data[:,2:end])
# Select features and create an index (or a database).
features = CellFishing.selectfeatures(counts, featurenames)
database = CellFishing.CellIndex(counts, features, metadata=cellnames)
# Save/load the database to/from a file (optional).
# CellFishing.save("database.cf", database)
# run(`gzip database.cf`) # or run(`zstd database.cf`)
# database = CellFishing.load("database.cf.gz")
# Load expression profiles of query cells.
data = CSV.read("query.txt", delim='\t')
cellnames = string.(names(data))
featurenames = string.(data[:,1])
counts = Matrix(data[:,2:end])
# Search the database for similar cells; k cells will be returned per query.
k = 10
neighbors = CellFishing.findneighbors(k, counts, featurenames, database)
# Write the neighboring cells to a file.
open("neighbors.tsv", "w") do file
println(file, join(["cell"; string.("n", 1:k)], '\t'))
for j in 1:length(cellnames)
print(file, cellnames[j])
for i in 1:k
print(file, '\t', database.metadata[neighbors.indexes[i,j]])
end
println(file)
end
end
不過嘛~ 說要用這個,目前俺手邊也還沒有資料,如果哪位大大手邊正巧有single-cell sequencing的資料可借來玩玩,還望有這機會可以借一步聊聊。

