前幾天雜七雜八地寫了很多東西,最主要的點就在於—生物資訊的分析本身通常都是由一堆別人開發好的工具加上我們自己寫的資料處理scripts所組成(我是指如果不討論到演算法開發的部份)。以這個前提,我們實際上要做的事情大多數的情況下都很不一定,除非是一個相當成熟的issue(像是從Microarray、RNA-seq甚至是Proteomics等資料去挖表現量有顯著差異的基因、蛋白質,再接著搞functional analysis),不然生物資訊領域目前還真的沒什麼解法能夠one size fits all。
在生物資訊的分析當中,個人覺得shell指令的重要性完全不亞於我們所學的其他程式語言。不管是為了讓程式碼看起來更簡潔或是為了加速或節省資源,pipe(管線)在我們常寫的scripts裡面,絕對扮演著一個舉足輕重的角色。在shell底下使用pipe當然不是什麼問題,但在Julia底下要怎麼使用這樣的功能呢?這就是我今天想要介紹的東西:
執行外部命令
Backtick notation:根據Julia官方文件裡面所寫的,Julia為了讓使用者能夠使用到外部命令,從而借助了backtick這種標記來標注我們現在給的一段字串是一種Cmd類的物件而不是一個String類的物件,這點與其他語言也很不一樣,其他語言看到這種標記就會去執行裡面的指令,但Julia只是把這段字串變成一個類別物件(見下圖)。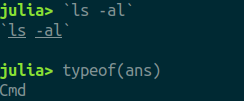
另外要保存外部命令執行後輸出的結果得另外處理,Julia預設是直接將結果輸出到STDOUT跟STDERR中。
Julia執行外部命令要透過run()這個函數,但實際上Julia並不是將這段命令丟給Shell處理,而是自己內部parse過之後另外開一個process去跑的(這邊我的理解可能有點問題,歡迎比較了解的人能給我一點comments)。
下面我們來看一個簡單的例子:
saysomething = "Hello world"
mycmd = `echo $saysomething`
run(mycmd)
串連命令 pipeline()
Shell裡面,我們很習慣地使用|這個符號來串接各種命令,在Julia裡頭,則是使用pipeline()這個函數: run(pipeline(`cut -d: -f3 /etc/passwd`, `sort -n`, `tail -n5`))
run(pipeline(`echo world` & `echo hello`, `sort`))
STDOUT丟出結果的,因此我們最終會將這些管線命令的輸出透過重新導向輸出到某個檔案裡,在Julia裡頭我們可以這麼做:pipeline(`do_work`, stdout=pipeline(`sort`, "out.txt"), stderr="errs.txt")
readlines()及readstring(),但個人這兩天在測試卻沒看到v1.0.0的Base這個package中有readstring()這個函數,不知道有沒有人能夠解答?datatable_file = "FullDataTable_filtered.txt"
headers = split(readlines(`head -n 1 $datatable_file`)[1], "\t")
@async:writer = @async writeall(process, "data")
reader = @async do_compute(readstring(process))
wait(process)
fetch(reader)
呼~ 今天就先到這邊啦! 明天再戰~

