Nextflow是一種基於Groovy語言寫成的workflow framework。Workflow framework對我們這些搞生物資訊的人來說是個很方便的工具,以往大家在分析各種data時,尤其是XX體(基因、蛋白或是微生物)相關的raw data時,往往得先對這些原始數據進行處理、過濾甚至檔案轉換等步驟,而這些步驟又通常是參考一些文獻或是產生那些原始資料用的設備商所提供的白皮書上面所說的方式來實做的。為了把這些步驟一個個串接起來,在過去,我們會選擇使用shell script或是perl之類的script language來把這些工作自動化。在當時,分析流程還沒這麼複雜的時候,可能還沒什麼問題,但隨著技術的進步,光是要做個簡單的分析就得用上好幾套不同的工具輪番上陣。因此,隨之而來的版本號問題、研究可重複性問題等也就成了當今生物資訊領域急需解決的一個難題。於是這些專門搭配資料處理流程的框架工具,也就在這幾年一一冒出來了。我自己目前知道的workflow framework就有以下這幾套 參考文獻:
在這麼多的選擇當中,為什麼我會選擇Nextflow來用呢?主要是去年在ISMB 2017這場國際研討會上有個BOSC開的section都在介紹專門用來開發bioinformatics pipleine用的workflow framework,當時一見Nextflow簡直驚為天人啊!!!
誠如官網上所說的:
conda、docker甚至是雲端環境,還可用Environment Module來管理工具的版本Channel以stream方式傳輸$ curl -s https://get.nextflow.io | bash
裝完馬上來測試一下
$ ./nextflow run hello
#!/usr/bin/env nextflow
people = ['Ronaldo', 'Maradona', 'Henry', 'Messi']
process runJulia {
input:
val name from people
output:
stdout results
script:
"""
#!/usr/bin/env julia
greetname = "$name"
println("Hello, ", greetname)
"""
}
執行後可以看到: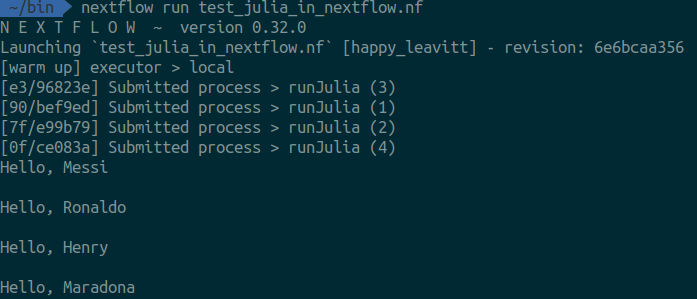
是不是很方便呢? 以後有機會再來跟大家分享Nextflow相關的東西。
補個 KNIME,open source 軟體,因為有圖形介面,一般人應該?比較會操作。
本身是一般商業的統計分析工具,有一點生物資訊的套件,也可串接 R、python。
在質譜、蛋白質體的分析上則有 openMS 套件。
質譜的資料目前我自己倒是沒什麼機會碰到~ 大大要來分享一下嗎? 我是說到社群XD
好哦,已加fb社群。如果有我可以分享的部份
歡迎大大! 一月份有空來分享一下嗎? <_ _> 感恩

