最新版本請見:https://chenhsieh.com/post/bioinfo/09-trimmomatic/
fastq 檔案中存放的資料包括序列及其對應的品質,雙端定序的結果分別存放在結尾標記 -1 和 -2 的檔案中。必須將定序品質不佳的鹼基剔除掉,才可以進行後續的分析。
Trimmomatic 是一款 java 撰寫的跨平台軟體,透過終端機指令提供參數來使用。

點選最新版本的 binary 即可,該頁面下方就有快速上手教學
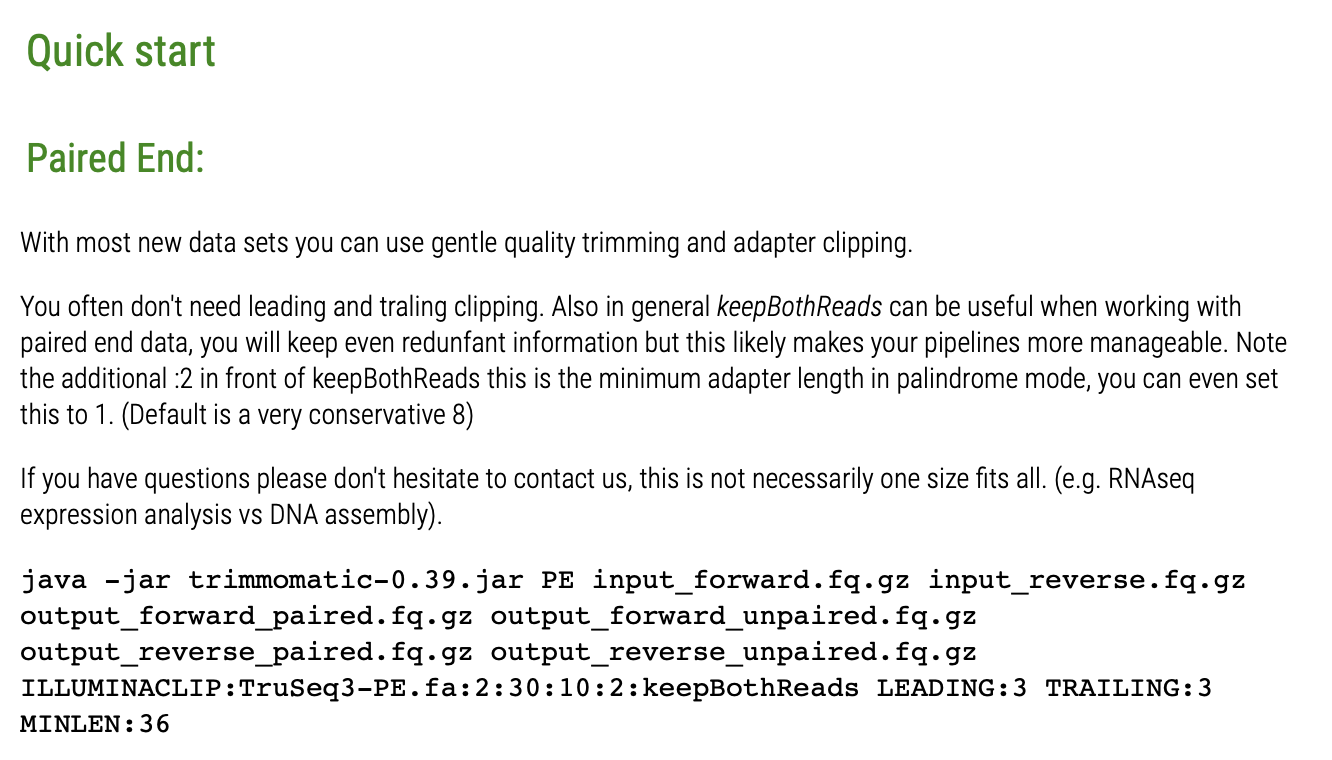
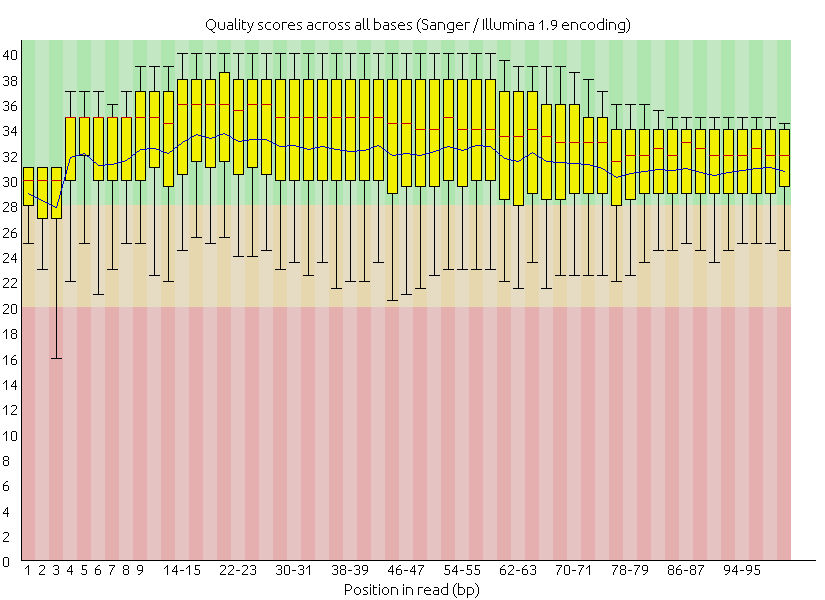
先來一段簡單的示範,下一段落才有實際手把手操作。原本品質差的序列以 FastQC 視覺化品質之盒方圖,可見許多位於末端的鹼基之定序品質已經來到紅色的不良範圍
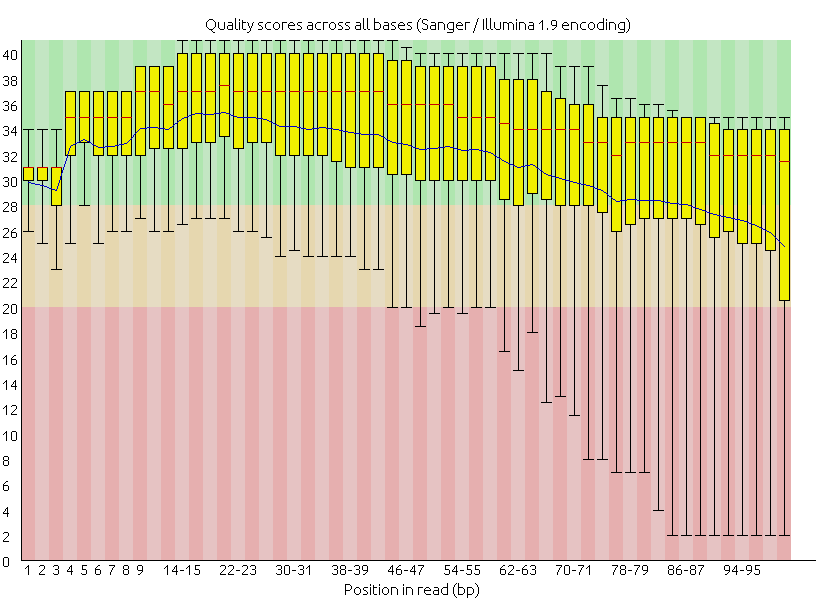
我們修剪的是雙端定序檔案,同一對序列來自同一條 DNA 的兩端,中間間隔 100 至 300 不等的未知序列。因此每一對序列的修剪過程與產出檔案如下:
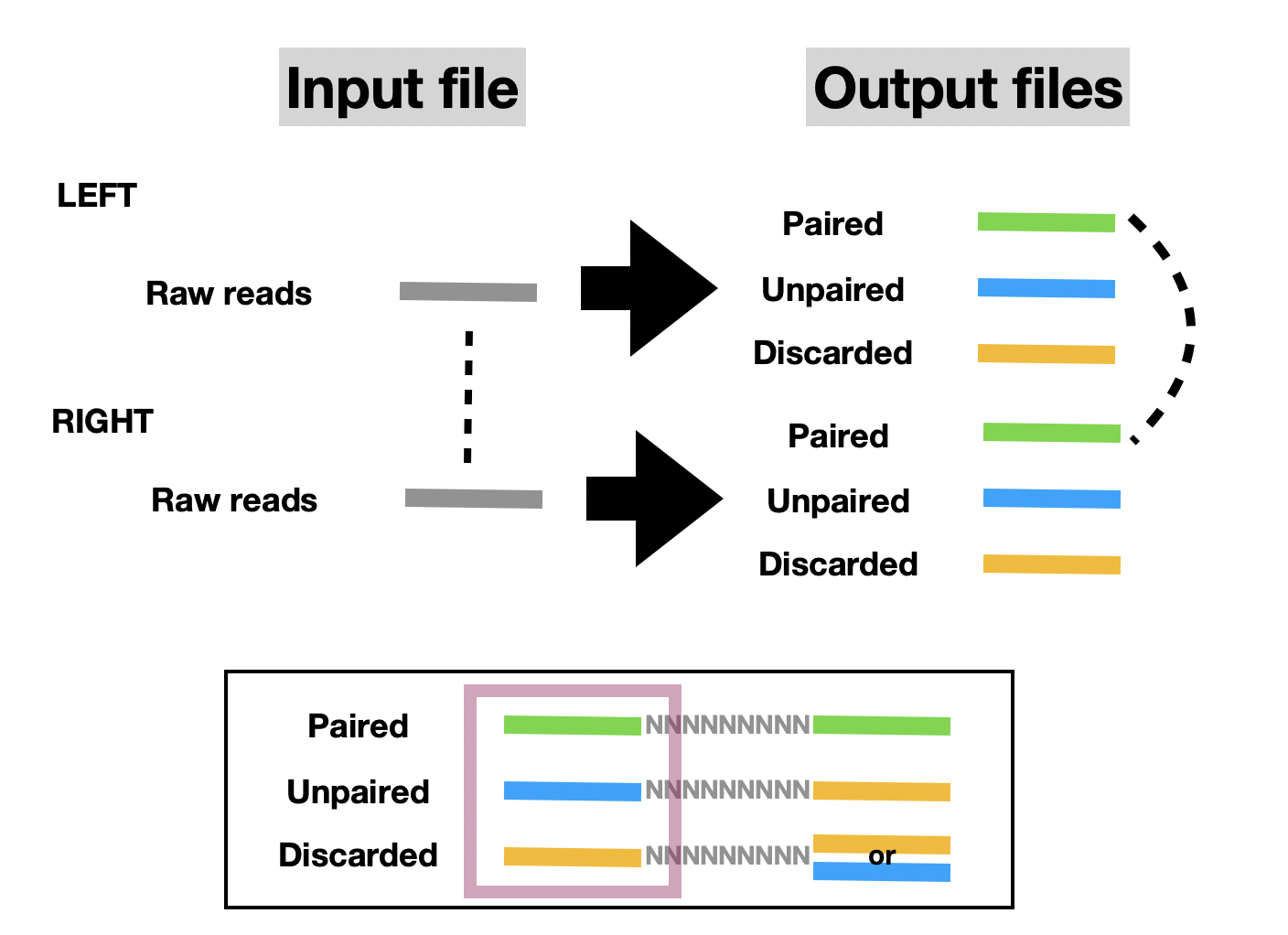
也就是說,輸入一對 (兩組) 序列檔案,會跑出一對+兩組 (共四組) 序列檔案,一對成對與兩組非成對。
在修剪後,不論是成對或非成對的修剪結果,品質都來到了安全的綠色區塊
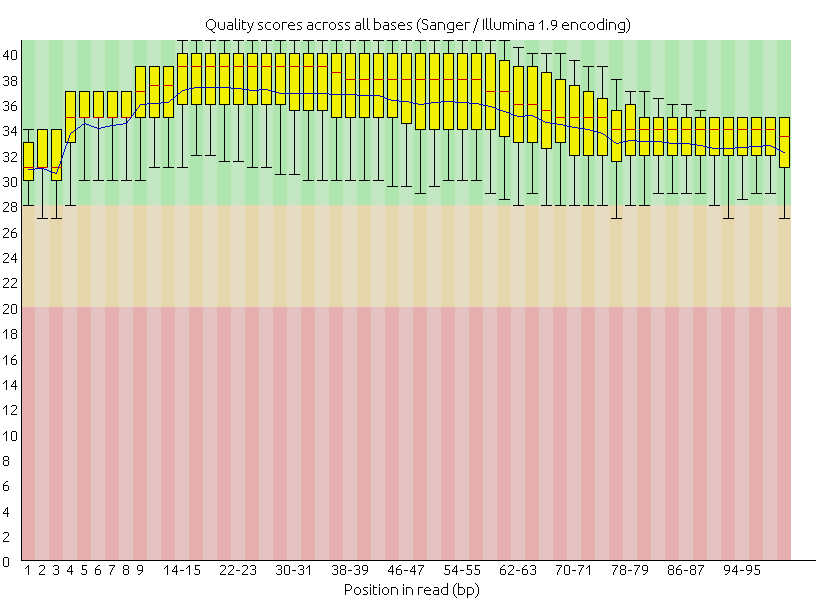
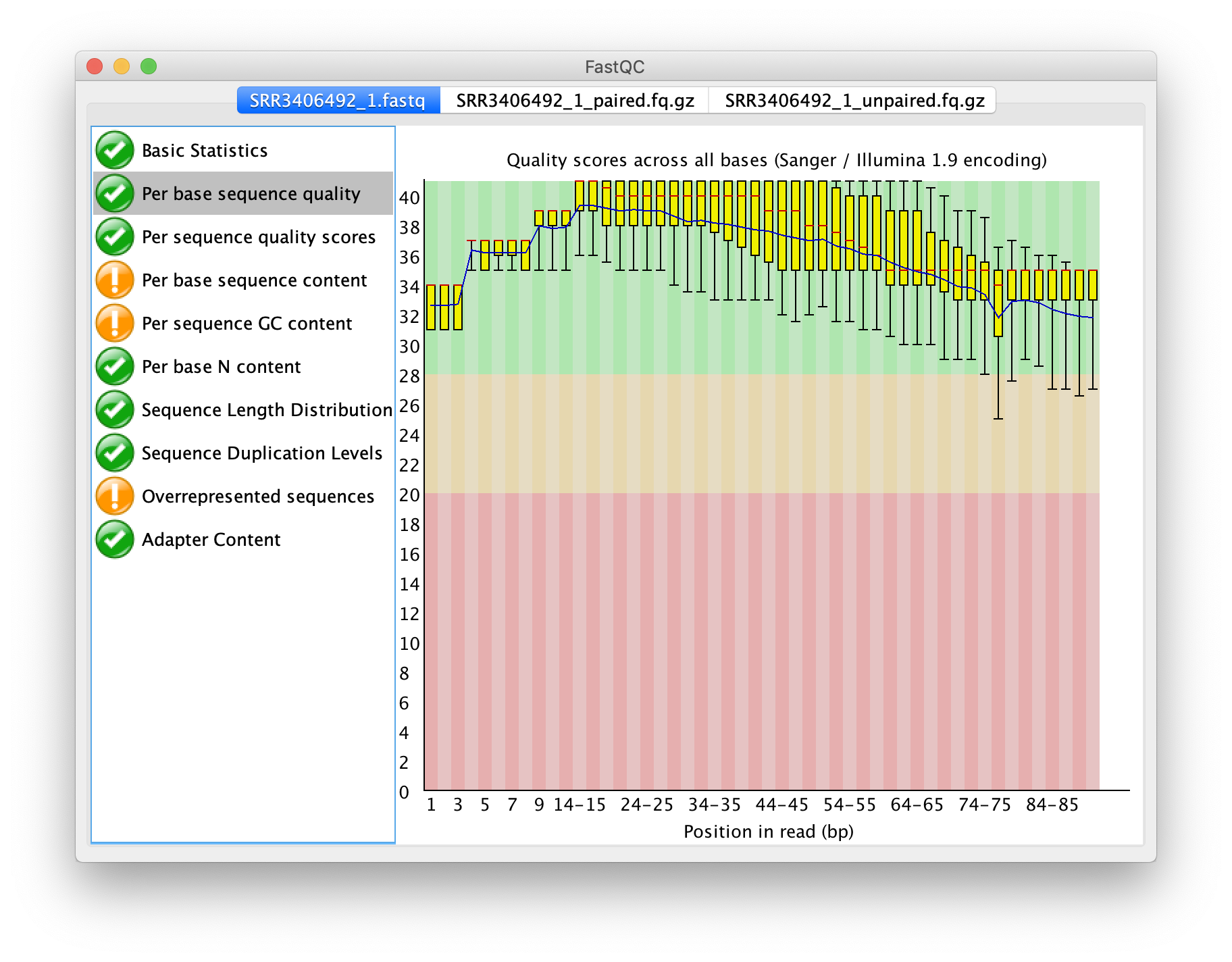
輸出之成對序列品質之盒方圖
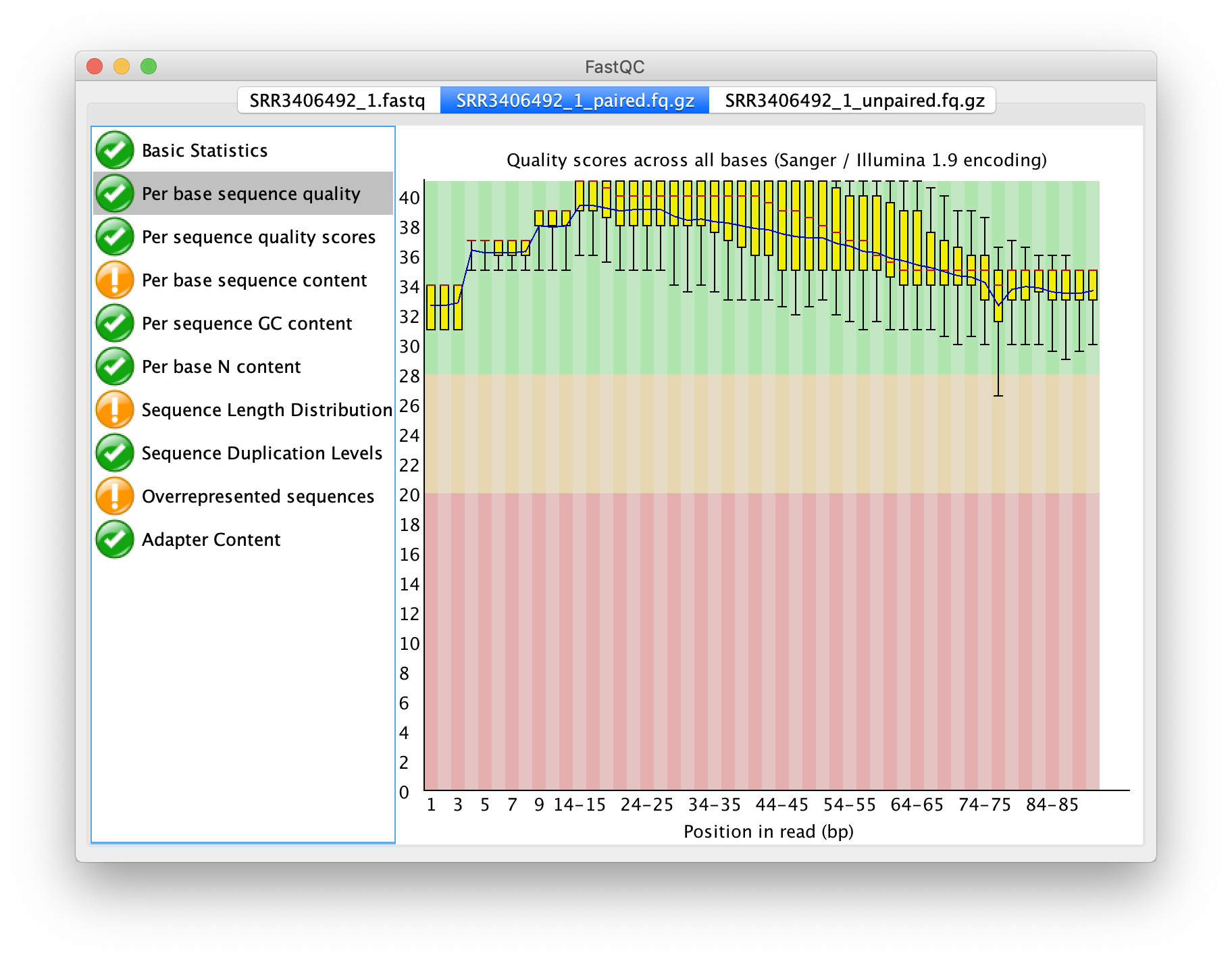
輸出之非成對序列品質之盒方圖
手癢想試試看吧?一樣以線上資料練習,以下方指令下載,僅有五千條讀序 (spot)
fastq-dump -X 5000 --split-files SRR3406492
這個練習中以 SRA 上的資料示範,但是這是已經被修剪過的檔案,因此品質極佳,輸出結果僅供參考。
以下列的預設指令修剪,每個參數所代表的意思,以淺綠底色標注在其旁邊

指令複製區!在 trimmomatic-0.39.jar 之目錄中執行,要修剪的原始檔案放在其上一層,記得注意軟體版本名稱、輸入輸出檔案之名稱要自行更改:
java -jar trimmomatic-0.39.jar PE -phred33 ./../SRR3406492_1.fastq ./../SRR3406492_2.fastq SRR3406492_1_paired.fq.gz SRR3406492_1_unpaired.fq.gz SRR3406492_2_paired.fq.gz SRR3406492_2_unpaired.fq.gz ILLUMINACLIP:./adapters/TruSeq3-PE.fa:2:30:10 LEADING:3 TRAILING:3 SLIDINGWINDOW:4:15 MINLEN:36
共輸出四個檔案,操作者要自行命名。執行成功會得到如下的回傳結果,五千條序列中有 98.84% 都被留下來了!
(base) Benjamins-Device:Trimmomatic-0.39 bendjamin101001$ java -jar trimmomatic-0.39.jar PE -phred33 ./../SRR3406492_1.fastq ./../SRR3406492_2.fastq SRR3406492_1_paired.fq.gz SRR3406492_1_unpaired.fq.gz SRR3406492_2_paired.fq.gz SRR3406492_2_unpaired.fq.gz ILLUMINACLIP:./adapters/TruSeq3-PE.fa:2:30:10 LEADING:3 TRAILING:3 SLIDINGWINDOW:4:15 MINLEN:36
TrimmomaticPE: Started with arguments:
-phred33 ./../SRR3406492_1.fastq ./../SRR3406492_2.fastq SRR3406492_1_paired.fq.gz SRR3406492_1_unpaired.fq.gz SRR3406492_2_paired.fq.gz SRR3406492_2_unpaired.fq.gz ILLUMINACLIP:./adapters/TruSeq3-PE.fa:2:30:10 LEADING:3 TRAILING:3 SLIDINGWINDOW:4:15 MINLEN:36
Multiple cores found: Using 4 threads
Using PrefixPair: 'TACACTCTTTCCCTACACGACGCTCTTCCGATCT' and 'GTGACTGGAGTTCAGACGTGTGCTCTTCCGATCT'
ILLUMINACLIP: Using 1 prefix pairs, 0 forward/reverse sequences, 0 forward only sequences, 0 reverse only sequences
Input Read Pairs: 5000 Both Surviving: 4942 (98.84%) Forward Only Surviving: 45 (0.90%) Reverse Only Surviving: 11 (0.22%) Dropped: 2 (0.04%)
TrimmomaticPE: Completed successfully
再次以 FastQC 檢測序列品質,成對之輸出,僅比原本的狀況提升一點點
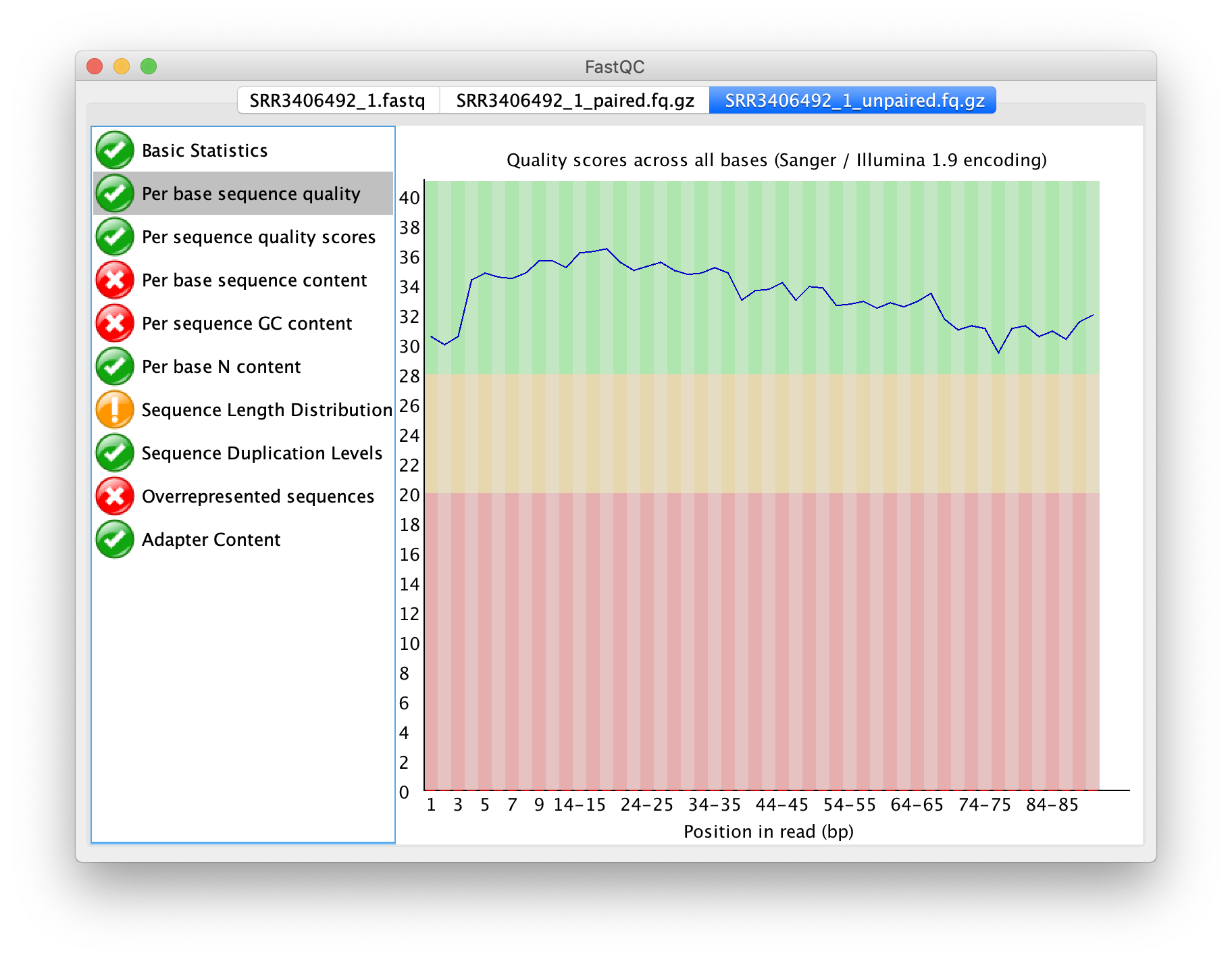
非成對之輸出
以上就是 Trimmomatic 的使用紀錄,如果有任何安裝與使用上的問題,需要中文支援,歡迎在下方留言~
USADELLAB.org - Trimmomatic: A flexible read trimming tool for Illumina NGS data
Trim Reads with Trimmomatic - v0.36 | KBase App
關於作者
謝晨 (Chen Hsieh),臺大園藝暨景觀學系研究所碩士。讀碩士前的興趣是懷著寫點程式妄圖解決農業問題的夢想參加比賽,拿了幾個黑客松與 Open Data 創新應用競賽的獎,卻都沒有勇氣將項目經營下去;研究所期間的興趣轉換成讀學術期刊的出刊電子報。靠著這些興趣當選 107 學年的臺大優秀青年,畢業後在農場旁的研究館辦公室寫點東西,希望可以跟世界分享生物資訊與園藝的樂趣!
感謝選擇匿名的朋友協助校閱初稿與提供意見,也敬請各位讀者不吝指教!
• Website: ChenHsieh.com

