演化的基本假設之一就是不同的物種或不同的基因家族成員可能具有共同的祖先,也就是同源(homology) 的概念。我們將序列相似度高的基因歸為一群,假設他們同源。序列相似度需要實際的統計方式來定義,也就是透過 BLAST (Basic Local Alignment Search Tool)、FASTA、HMMER 等等序列比對軟體輸出的得分來衡量。序列比對軟體比對的對象可以是序列 (sequence) 本身,也可以是用 profile。profile 在此翻譯成輪廓,代表序列的輪廓,由一組相似的序列為材料,比如同一個基因家族的同一個 domain,就可以透過 HMM 的過程來建立一個基於機率的輪廓。
直接來個例子~
現在想要找出苦瓜中所有的 NAC 轉錄因子,有三種策略:
pfam 已經搜集了所有有發表的 domain 的序列,供大家搜尋、檢閱、下載,下載的格式是 Stockholm,其中包含了已經初步比對過的序列。Stockholm 檔案格式如下
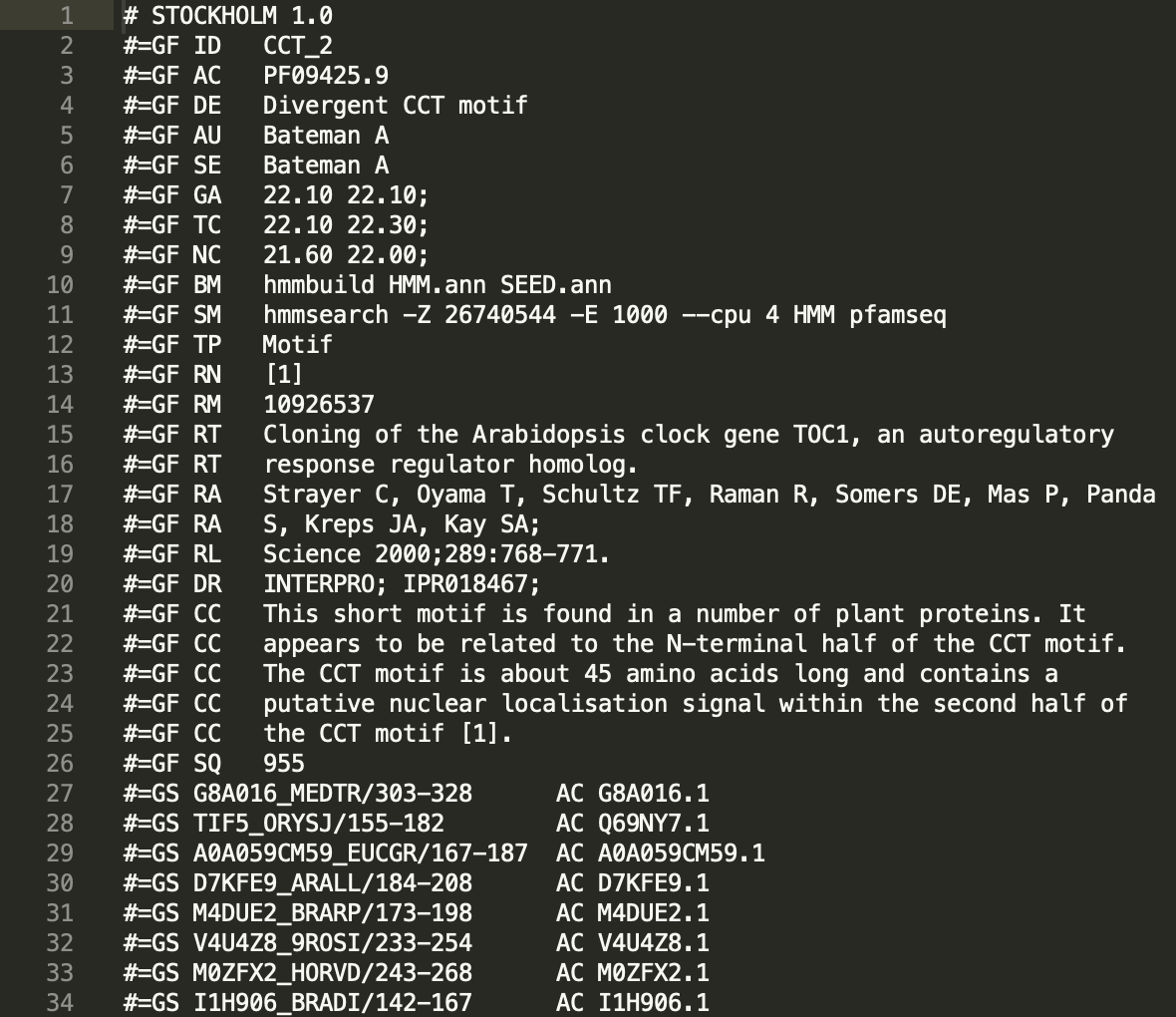
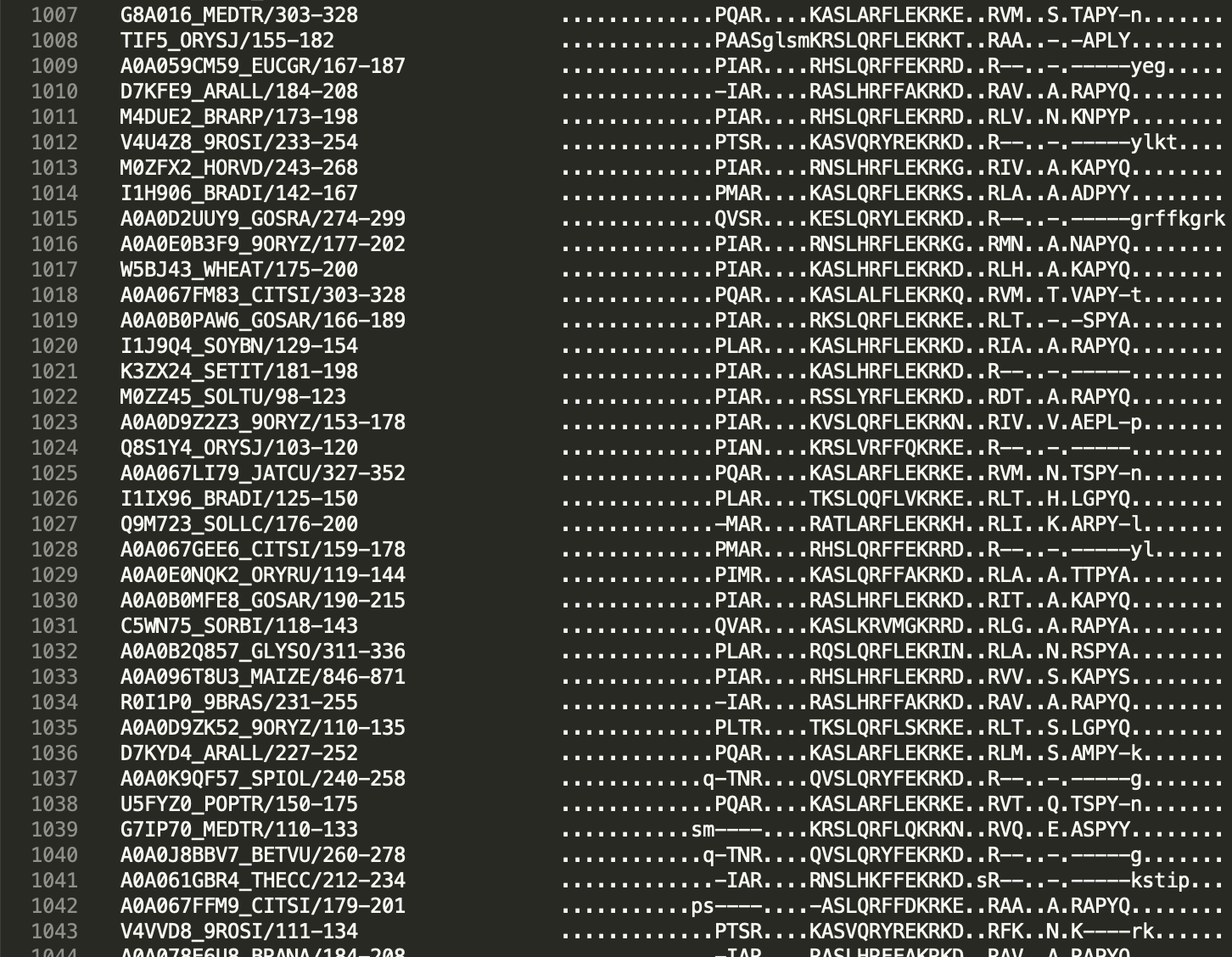
假設我們下載了一個叫做 cct 的比對檔案,檔案名稱為 cct.sto,下一步就要使用 hmmer 針對這個比對的檔案建立一個 cct 的 hmm profile,檔案名稱為 cct.hmm
hmmbuild cct.hmm cct.sto
.hmm 檔案的格式如下,其中數值為由序列比對檔案運算之機率
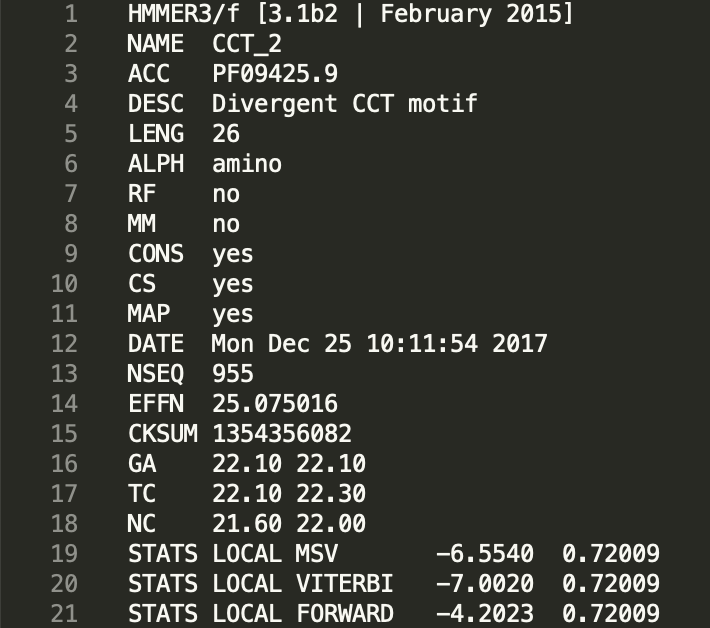
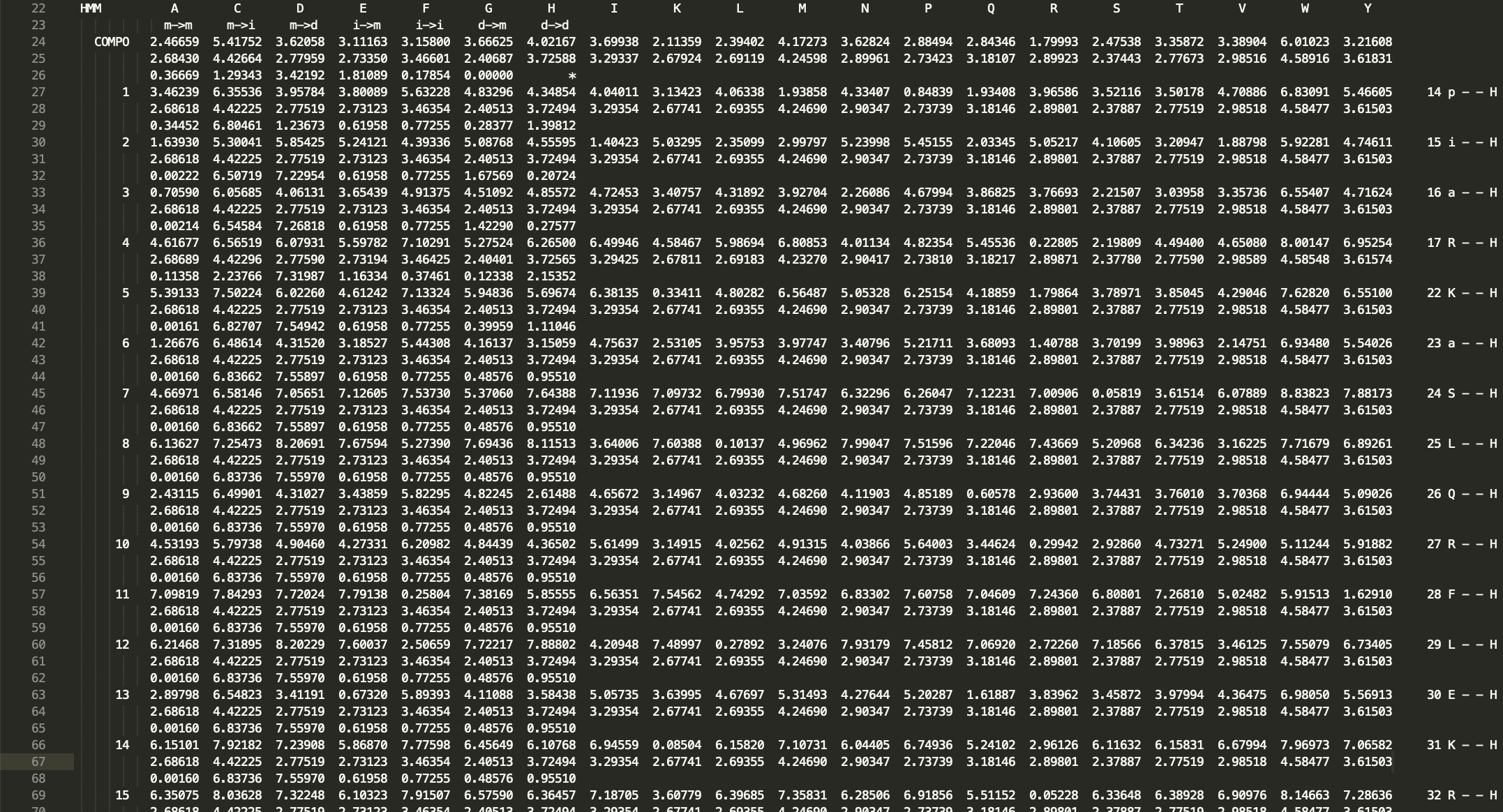
之後再使用下列指令,對 transcriptome.fa 的所有序列,以 cct.hmm 搜尋,找出符合其輪廓的序列
hmmsearch cct.hmm transcriptome.fa
輸出的檔案格式包括標題、入選的序列、以及條入選序列與 profile 的比對結果
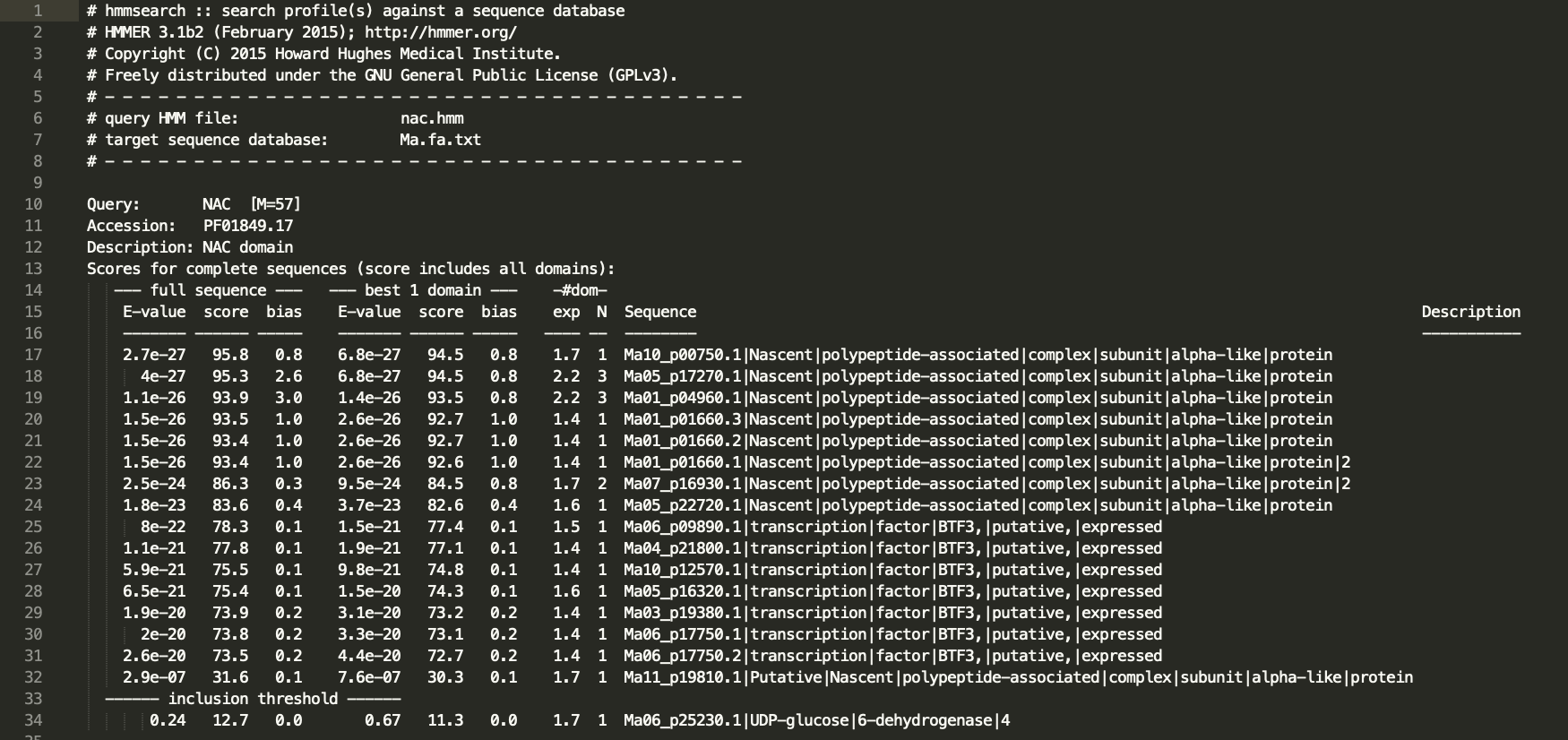
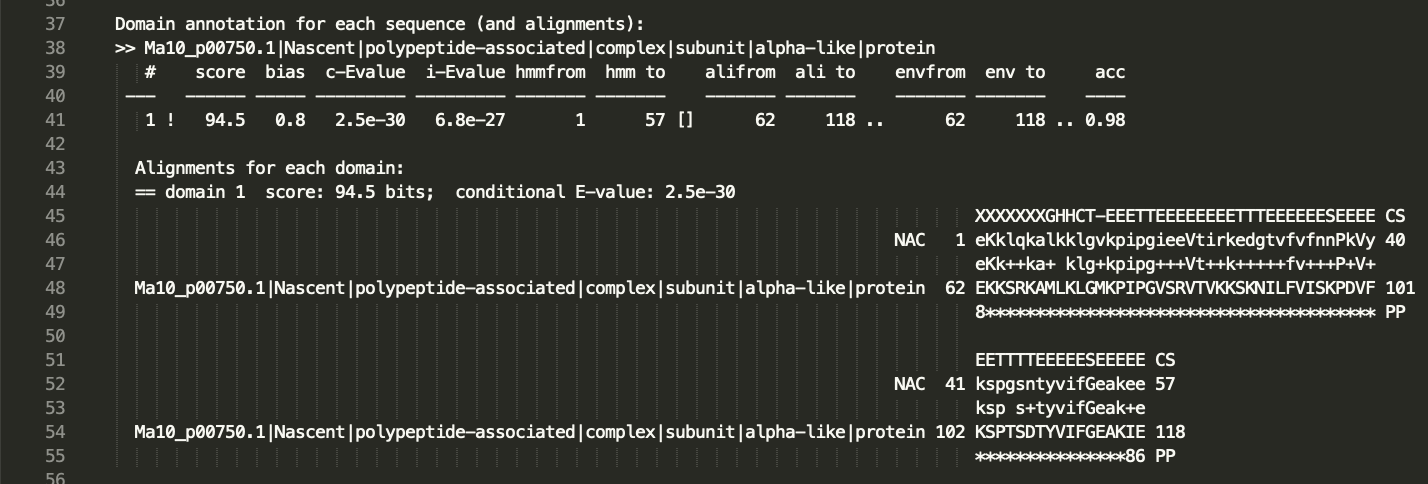
最後輸出的檔案結果其實跟 BLAST 的結果很像,就是一些候選的序列跟他們兩條比對夠相似的片段的連連看,但是背後原理大不同。至於實際操作上來說,如果使用 BLAST 的話,那就是序列對序列的搜尋,胡瓜中有一百條 NAC 的話,我們就要把一百條 NAC 都抓下來每一個都對苦瓜轉錄體執行一次 BLAST,其中有些不重複的輸出結果又該保留就會變成一場災難。相較之下, HMMER 是以一群相似的序列來建立機率 profile,更可以代表這一整群參考的序列先祖的共同的狀態,以一個 profile 去搜尋一套轉錄體,輸出的結果就可以很確定地供下游分析啦!
關於作者
謝晨 (Chen Hsieh),臺大園藝暨景觀學系研究所碩士。讀碩士前的興趣是懷著寫點程式妄圖解決農業問題的夢想參加比賽,拿了幾個黑客松與 Open Data 創新應用競賽的獎,卻都沒有勇氣將項目經營下去;研究所期間的興趣轉換成讀學術期刊的出刊電子報。靠著這些興趣當選 107 學年的臺大優秀青年,畢業後卻成了無業的實驗室居民。現在在農場旁的研究館辦公室寫點東西,希望可以跟世界分享生物資訊與園藝的樂趣!
感謝選擇匿名的朋友協助校閱初稿與提供意見,也敬請各位讀者不吝指教!

