
昨天的文章簡單地介紹了幾種偵測CNV的實驗方法及其原理,原本想繼續寫關於CNV-calling的東西,不過這幾天實在是休息不夠,狀態不是很好。這兩三天我打算陸續介紹幾個我自己所知的用來做CNV-calling的生物資訊方法,這些方法不限於使用在處理、分析哪一種實驗技術所產生的資料。主要的原因是這些方法各自有各自的優點及缺點,我們無法很絕對地說哪種方法是最好的。此外,在許多的研究當中,即便是使用了NGS的技術加上特定的CNV caller,最終它們仍會補上某個使用不同實驗技術平台所得到的結果作為驗證,不過對於這點,今年四月有篇發表在PLOS One上的文章就抱持著相當不同的觀點,有興趣的人可以自行看看。
在介紹CNV-calling的方法之前,我想我必須先介紹幾個專有名詞,一方面是讓比較熟的讀者幫我判斷我的認知對不對,一方面還可以幫同樣是新手的朋友建立所需的基本認知。
B allele frequency (aka. BAF):當初看到這個東西的時候只是大概理解成這個allele是homozygous及heterozygous的一種判斷頻率而已,但後來看到Biostars上面的討論,才發現這個東西遠比我想像中的還複雜一些。最早的A/B allele之分是從Illumina的晶片平台出來的,目的是為了可以讓使用者可以區分每一個allele是來自DNA的哪一股,確定了該probe的方向後,再區分在這一個allele上有哪些variants,有興趣了解的朋友請看Illumina的technical note。了解了這段歷史淵源後,再回來看BAF,把它理解成所謂的Allele frequency就對了(如果是AA的話其值是1,如果是BB則是0,如果是AB的話其值是0.5,見下圖)。
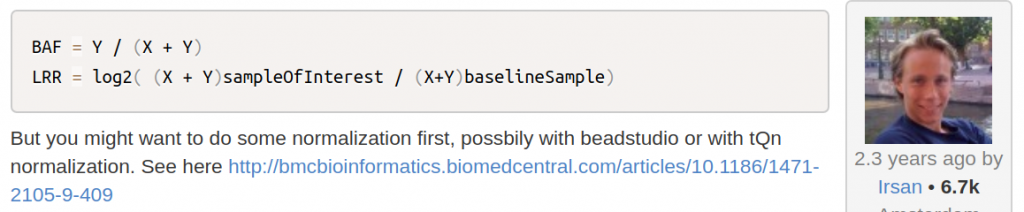
Log R ratio (aka. LRR): 經過歸一化後的信號強度觀測值與信號強度期望值的比例。這兩個值要怎麼求得呢? 來看一下Biostars上面給的簡單公式:
PennCNV是一個基於HMM演算法的工具,跟更早期同樣也是HMM-based的方法相比,它還可以引入家族遺傳資訊,透過貝氏定理,推論出posteriori CNV validation及CNV boundary mapping的機率,提昇CNV-calling的精準度。作者以{r_i, b_i, z_i}分別表示第i個SNP的LRR、BAF以及CNV state,這裡CNV state是hidden state。

假設LRR及BAF與CNV state無關,在已知CNV state的條件下,觀察到這樣的LRR及BAF值的機率就可以表示成:

LRR的emission probability可以表示成:

BAF的emission probability比較複雜,主要是因為考慮到每個CNV state都分別可以對應到多個genotypes:

在這裡 
不同CNV state之間的狀態轉移機率可表示成:

最終的HMM model可以寫成:

整個演算法的邏輯以這張圖來表示:

https://github.com/WGLab/PennCNV

 iThome鐵人賽
iThome鐵人賽