
所以今天就不意外地要介紹一下目前用來分析NGS資料當中CNV的方法了!根據Computational tools for copy number variation (CNV) detection using next-generation sequencing data: features and perspectives這篇論文所提到的,一般來說,用在NGS data上找CNV的方法大概可以粗分為五大類:
Paired-end mapping (PEM): 這種方法是根據檢查paired-end reads之間的距離(inner distance)是否顯著與平均的距離不同,來判斷CNV。Split read (SR): 這種方法利用每一對read pair的incompletely mapping來判斷是否有small CNVs。Read depth (RD):顧名思義就是利用了比較比對上每一個genomic region上面的reads數量來做判斷。De novo assembly of a genome (AS):這個方法就概念上來說我還蠻喜歡的,它先將所有的reads透過de novo assembly組裝成contigs之後來跟reference genome做比較,來判斷CNVs。Combination of the above approaches (CB):主要是同時運用了PEM跟RD兩種方法來判斷CNVs。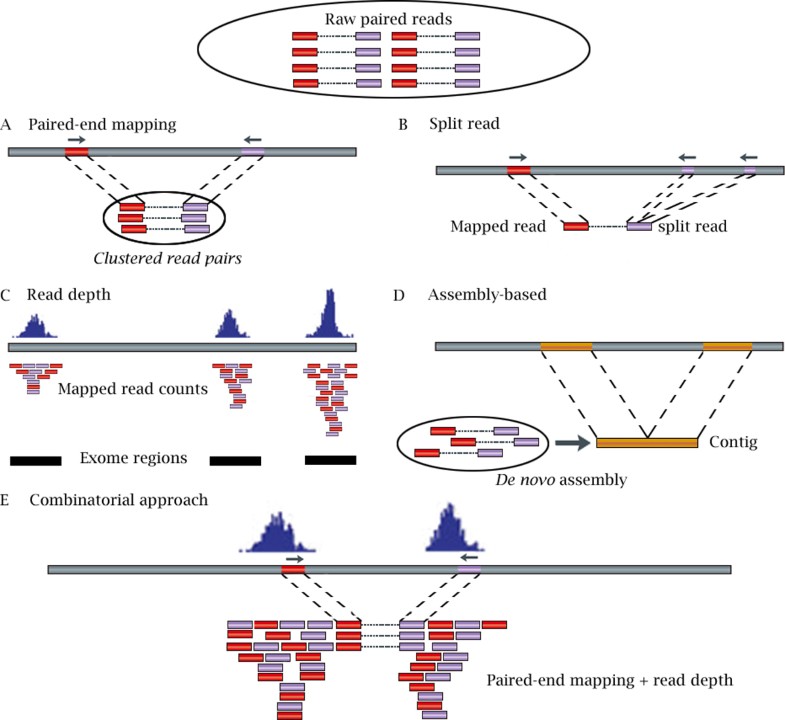
但是就我自己看到目前為止,絕大部分的新工具或是新方法幾乎都是read-depth-based不過我還是順便附上一下我自己在寫這一篇的時候所蒐集到的CNV tools資訊連結,附帶一提的是根據今年發表在American Journal of Human Genetics的一篇文章表示,目前大約有超過五十種不同的CNV-calling方法,其中被引用最多的一個也不過只佔了不到12%的引用量,加上這些不同方法所得到的一致性不是很高,因此真要說找一個極具代表性的工具還真的是沒辦法。
目前文獻中,一般比較推薦的分析流程如下:


 iThome鐵人賽
iThome鐵人賽