前一回我們探討了 NGS 從 DNA 樣本到上機 (上 Illumina 機器定序),
今天找找前人上機後的資料,究竟都去哪裡了~
如果已經有一批定序資料下載好等著你練習了,那麼就往下看下一篇XD
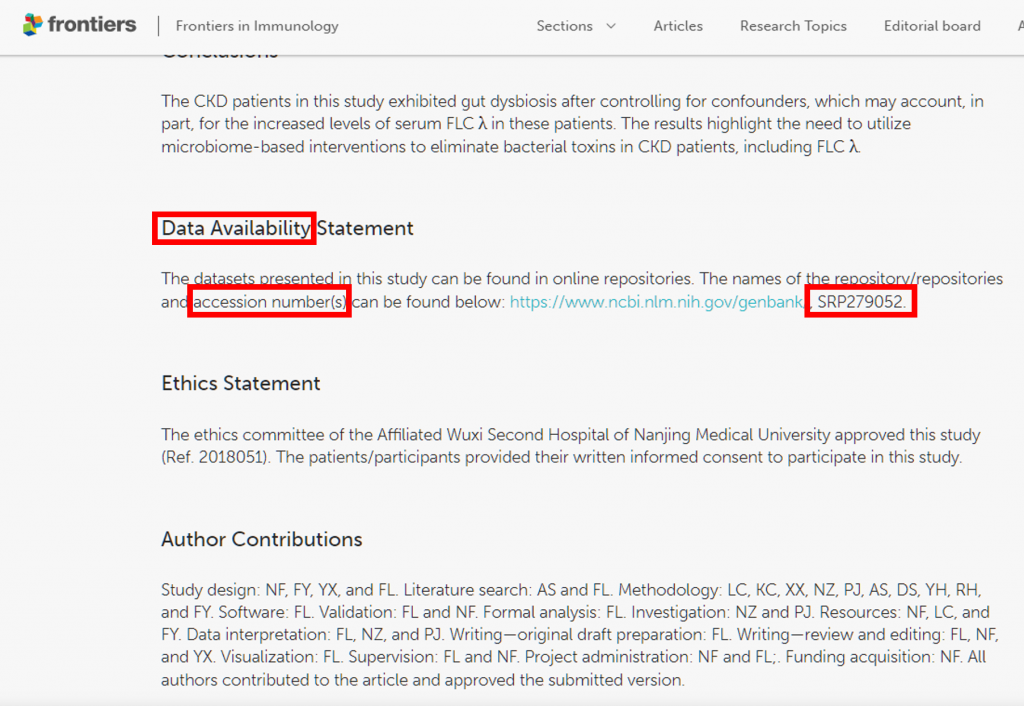
SRA (Sequence Read Archive) 資料庫存放全世界研究的定序資料,
基本上要發有關定序資料分析結果的 Paper 通常都會要求上傳到這兒,
包含 RNA seq、Single cell sequencing、Whole genome sequencing、Metagenomic(16S)等通常-->就代表很多人不乖
首先,得先在 Paper 中拿到一串 accession number,
有上傳到 SRA 的 Paper 則會在文中以 :
Data Availability、repositories、accession number、accession SRPXXXXXX 關鍵字出現,
範例1 : (Yang, Tzu-Wei, et al., 2019)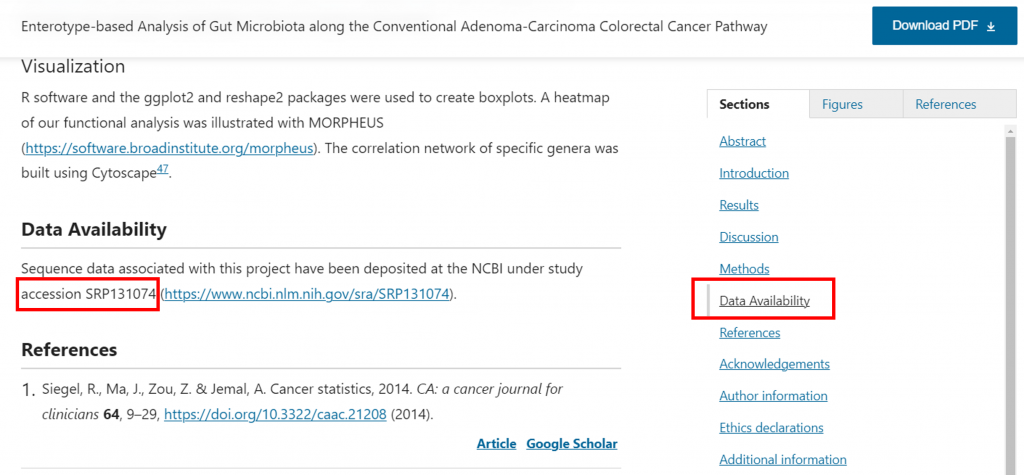
以範例1來說,
帶著 SRP131074 餵給 SRA Run Selector,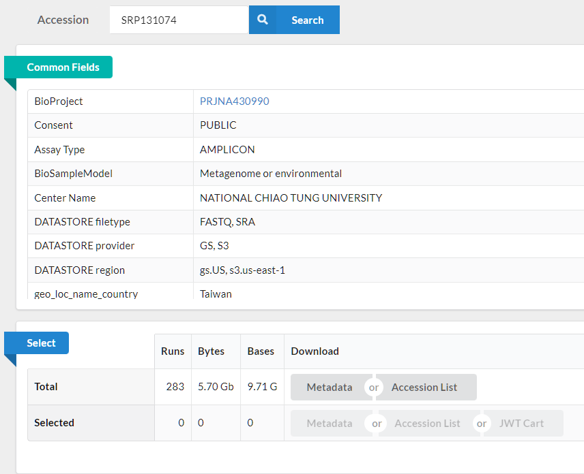
在Common Fields 中確認基本資訊是否與文獻提及的內容相符 (檢體類型、序列類型等),
通常我還會確認是否有 Platform : ILLUMINA、LibraryLayout : PAIRED、metagenome 等關鍵字。
往下滑會看到表格,
從 Library Name、Sample Name推敲每個序列檔案代表的資訊,
通常會與 Paper 中分組的代號相關,好心一點會放在文獻的 Supplementary Material,實務上很多都寫得不清不楚,所以需要一點推敲
在這裡我勾擇CC,代表已經發展成大腸癌的患者檢體(Colorectal Cancer),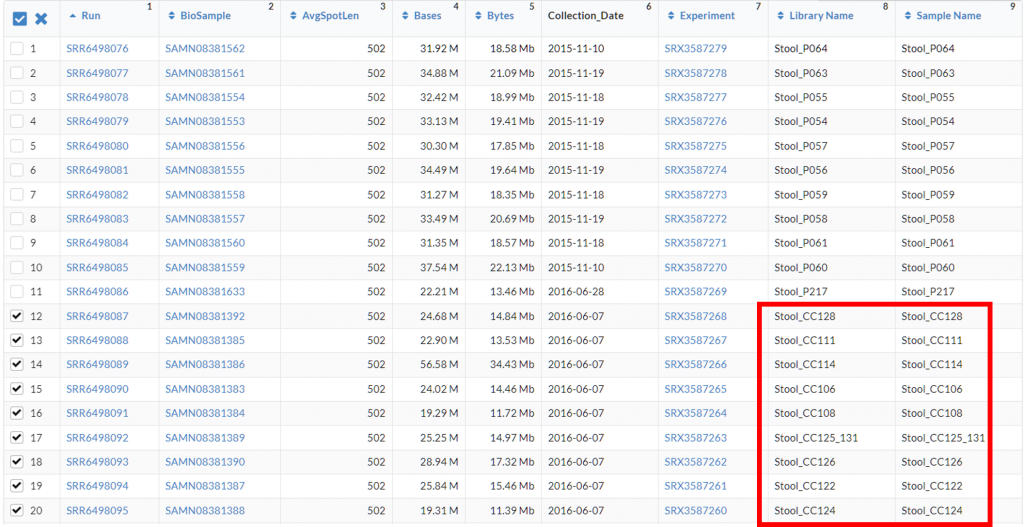
勾選後,可以往上滑點 Accession List 下載每個序列檔案的代號,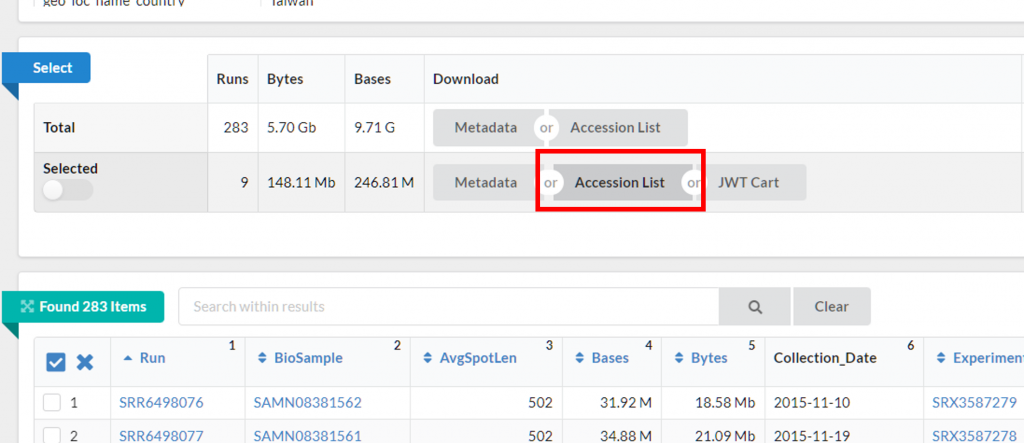
後續 SRA Toolkit 會根據 Accession List 檔案抓取資料,
偷偷點開下載的檔案,其實很單純,就是剛剛勾選的序列檔案代號。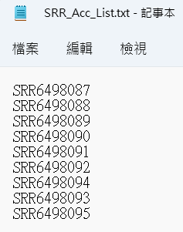
現在帶著 Accession List 要去 Linux 準備下載序列資料啦!
16S rRNA 序列檔案大小雖然較 RNA seq 小,
但是樣本數一多還是很可觀的,
SRA Toolkit 提供使用者可以快速下載 NCBI SRA 資料庫上的序列資料,
會依照你給的 Accession List 到 SRA database 下載資料。
但是對於一次需要下載數十個甚至數百個定序資料分析,
就需要 Python bioinfokit 提供批量下載的功能。
16S的資料分析往往需要好幾十個以上的樣本菌相比較,
才能看出整體菌相差異性。
下列流程是預設使用者使用 Linux,並已安裝完 Anaconda 或 Miniconda
先用 conda 創造一個虛擬環境,避免跟原本的你使用的 python 環境打架
可以把 conda 想成一種包含 Python 與各類幫助分析套件的軟體,
在 conda 建立虛擬環境(就像是沙盒),可以幫助我們從事不同的分析使軟體彼此不衝突,
例如 : 下載序列軟體放在一個沙盒、分析序列軟體放在另一個沙盒。
conda create --name SRA_download python==3.8
會顯示的畫面 (以miniconda為例) :
[XXXXXX@localhost]$ conda create --name SRA_download
Collecting package metadata (current_repodata.json): done
Solving environment: done
## Package Plan ##
environment location: /home/XXXXXX/miniconda3/envs/SRA_download
Proceed ([y]/n)?
輸入 y 後 Enter,會顯示的畫面 :
Preparing transaction: done
Verifying transaction: done
Executing transaction: done
#
# To activate this environment, use
#
# $ conda activate SRA_download
#
# To deactivate an active environment, use
#
# $ conda deactivate
創造完之後,輸入以下指令進入SRA_download虛擬環境 :
conda activate SRA_download
成功的話會在你的名稱前面出現 (SRA_download) :
(SRA_download) [XXXXXX@localhost ~]$
安裝 bioinfokit python 套件
conda install -c bioconda bioinfokit
輸入 y 後 Enter,會經歷一段漫長的過程(?,直到看到這三行出現就對了~
...
Preparing transaction: done
Verifying transaction: done
Executing transaction: done
再裝這些套件
pip install NumPy scikit-learn seaborn pandas matplotlib SciPy matplotlib_venn adjustText statsmodels tabulate textwrap3
跑出 Successfully 的相關訊息就全部安裝完成拉~
'Successfully' built matplotlib_venn adjustText
Installing collected packages: textwrap3, pytz, threadpoolctl, tabulate, six, pyparsing, pillow, NumPy, kiwisolver, joblib, fonttools, cycler, SciPy, python-dateutil, patsy, packaging, scikit-learn, pandas, matplotlib, statsmodels, seaborn, matplotlib_venn, adjustText
'Successfully' installed NumPy-1.23.2 SciPy-1.9.1 adjustText-0.7.3 cycler-0.11.0 fonttools-4.37.1 joblib-1.1.0 kiwisolver-1.4.4 matplotlib-3.5.3 matplotlib_venn-0.11.7 packaging-21.3 pandas-1.4.4 patsy-0.5.2 pillow-9.2.0 pyparsing-3.0.9 python-dateutil-2.8.2 pytz 2022.2.1 scikit-learn-1.1.2 seaborn-0.11.2 six-1.16.0 statsmodels-0.13.2 tabulate-0.8.10 textwrap3-0.9.2 threadpoolctl-3.1.0
下載 3.0.0 版本 SRA Toolkit 壓縮安裝檔到本機
wget https://ftp-trace.ncbi.nlm.nih.gov/sra/sdk/3.0.0/sratoolkit.3.0.0-ubuntu64.tar.gz
解壓縮安裝檔
tar -zxvf sratoolkit.3.0.0-ubuntu64.tar.gz
你在的目錄會多一個sratoolkit.3.0.0-ubuntu64資料夾,就是主程式。
接著設定環境變數
export PATH=$PATH:$PWD/sratoolkit.3.0.0-ubuntu64/bin
vdb-config -i
會顯示的畫面 :
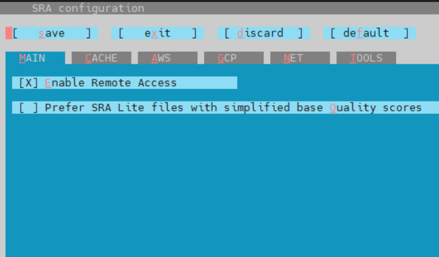
紅色底線的字母就代表進入需要按的按鍵
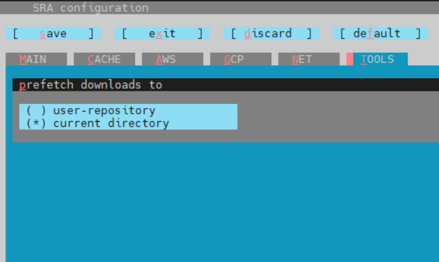
按 T 進入 TOOLS,再按 P ,上下鍵選擇 current directory,
再按 S 儲存,再按 o 代表 ok ,按兩次 Esc 即可離開。
你的目錄應該要有 :
最一開始下載的檔案SRR_Acc_List.txt,將他放到 Linux 目錄,
這邊也提供範例檔案包含六種序列
SRR_Acc_List.txt
SRR6498087
SRR6498088
SRR6498089
SRR6498090
SRR6498091
SRR6498092
接著我們製作一個 python 執行檔,檔名可自訂,教學方便叫做batch_download.py
內容如下 (可以在 Linux 以 vim 創建或是在 Windows / Mac 建好傳上去)
from bioinfokit.analys import fastq
fastq.sra_bd(file='SRR_Acc_List.txt', t=16, other_opts='--split-files' )
t=16,代表使用16核心file='SRR_Acc_List.txt',代表包含序列編號的檔案other_opts='--split-files',代表輸出的檔案要將Forward Reverse分別以_1, _2 表示
執行batch_download.py
python batch_download.py
會顯示的畫面 :
Donwloading SRR6498087
Donwloading SRR6498088
Donwloading SRR6498089
Donwloading SRR6498090
Donwloading SRR6498091
Donwloading SRR6498092
等到下一行再次出現,就代表完成啦!
[XXXXXX@localhost ~]$
可以 ll 查看目錄是否有該檔案
-rw-r--r-- 1 XXXXXX users 28171694 Aug 23 15:10 SRR6498087_1.fastq
-rw-r--r-- 1 XXXXXX users 28171694 Aug 23 15:10 SRR6498087_2.fastq
-rw-r--r-- 1 XXXXXX users 26139160 Aug 23 15:11 SRR6498088_1.fastq
-rw-r--r-- 1 XXXXXX users 26139160 Aug 23 15:11 SRR6498088_2.fastq
-rw-r--r-- 1 XXXXXX users 64697336 Aug 23 15:13 SRR6498089_1.fastq
-rw-r--r-- 1 XXXXXX users 64697336 Aug 23 15:13 SRR6498089_2.fastq
-rw-r--r-- 1 XXXXXX users 28171694 Aug 23 15:10 SRR6498090_1.fastq
-rw-r--r-- 1 XXXXXX users 28171694 Aug 23 15:10 SRR6498090_2.fastq
-rw-r--r-- 1 XXXXXX users 26139160 Aug 23 15:11 SRR6498091_1.fastq
-rw-r--r-- 1 XXXXXX users 26139160 Aug 23 15:11 SRR6498091_2.fastq
-rw-r--r-- 1 XXXXXX users 64697336 Aug 23 15:13 SRR6498092_1.fastq
-rw-r--r-- 1 XXXXXX users 64697336 Aug 23 15:13 SRR6498092_2.fastq
如果發現檔案名稱並非 .gz 副檔名結尾的壓縮檔,
記得將檔案做壓縮,避免後續分析報錯。
gzip *.fastq
到這裡都順利,可以將sratoolkit.3.0.0-ubuntu64.tar.gz殘餘的安裝檔刪除
rm sratoolkit.3.0.0-ubuntu64.tar.gz
只要啟動虛擬環境就可以從本篇的「下載小資料試試」,就能繼續分析新的資料。
conda activate SRA_download
而離開環境的方式 :
conda deactivate
就會看到前面的括號環境名稱消失,就是成功離開。
本篇文章同步刊載於科學毛怪部落格 PetSci Blog。