在前幾天知道了,
16S rRNA 菌相資訊的應用 [Day2]
16S rRNA 的二代 (NGS) 定序方式 [Day3]
16S rRNA 的定序後序列資料下載 [Day4]
如果要一言以蔽之本系列文就是 :
用 16S rRNA 做定序、分析獲得菌相資訊。
看來,我們剩下兩個名詞沒有介紹,
分別是被提了N次的 16S rRNA 還有 分析 :
✏️ 16S rRNA
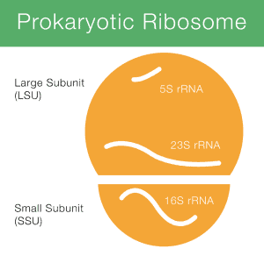
在原核細胞 (Prokaryotic) 核糖體 (ribosome) 含有 rRNA,
可以分成位於大次單元 (LSU) 的 5S 、23S rRNA (約2900 nt),
位於小次單元 (SSU) 的 16S rRNA (1542 nt)。
其中小次單元中的 16S rRNA 引起科學家高度興趣,
在物種演化過程,16S rRNA 在短短 1542 nt中,
不同物種間有些區段的序列出現差異,但也有一直保持不變的。
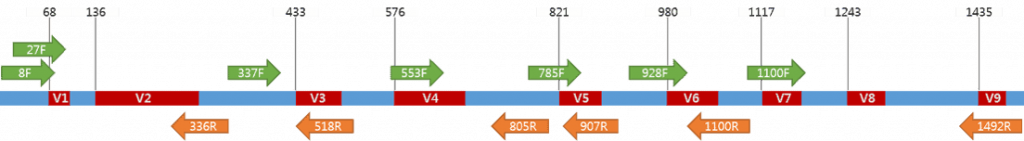
有差異的部分稱為 : 變異區 (Variable Region),分為V1~V9,上圖紅色區,
一直保持不變的稱為 : 保守區 (Conserved Regions),上圖藍色區。
如果你做過 PCR 可以想想若將 Primer 設計在保守區,
就可以夾到各種變異區的序列,而且還通用各物種(萬用Key),
是不是超棒的東東 !
於是科學家就拿著各種 Primer 試圖夾出各種菌種的變異區序列,
Primer 就是上圖的綠色(Forward)、橘色(Reverse)箭頭,
仔細看他們都從保守區出發。
夾到的片段再經由定序之後,
將其獨一無二的序列與物種階層連結,
這就是分生鑑種的技術。
✏️ 16S rRNA V3-V4 變異區雙尾定序 (V3-V4 region pair-end sequencing)
次世代定序雖然可以達到高通量(大量序列讀取),
但是每次讀取的長度有限 (單條超過 300 bp 品質就會急速下降),
所以無法快樂的從 V1 一路定到 V9 (>1400 bp)。
如今第三代定序解決了這個問題,之後會提及。
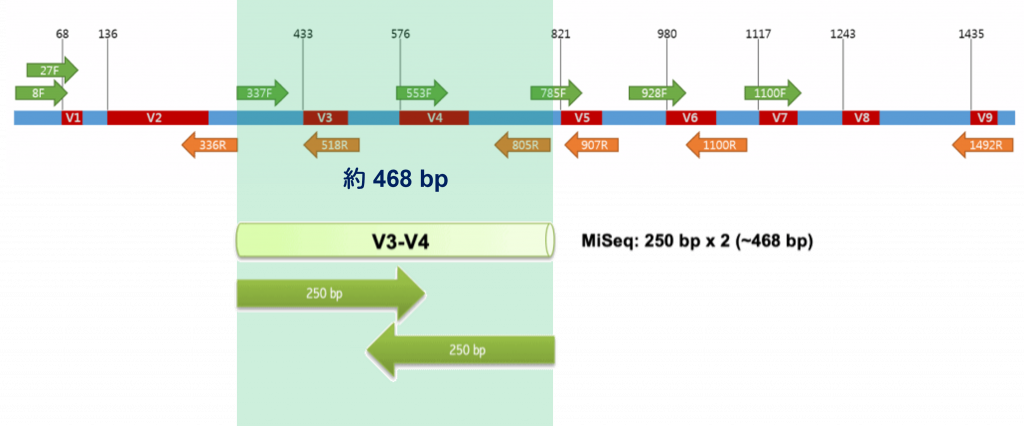
而科學家發現 V3-V4 區域 (約 470 bp) 在原核生物變異性很大,
在解析度 (resolution) 上足以辨別出各式各樣的菌,
但是超過 300 bp 品質就岌岌可危了 怎麼辦可好 ?
於是 Illumina Miseq 提出了 Pair-end sequencing 解決方案 :
採用了 337F 及 805R 兩種 Primer 從兩端夾攻,
這樣就獲得了兩條約 250 bp 的序列 (R1, R2),
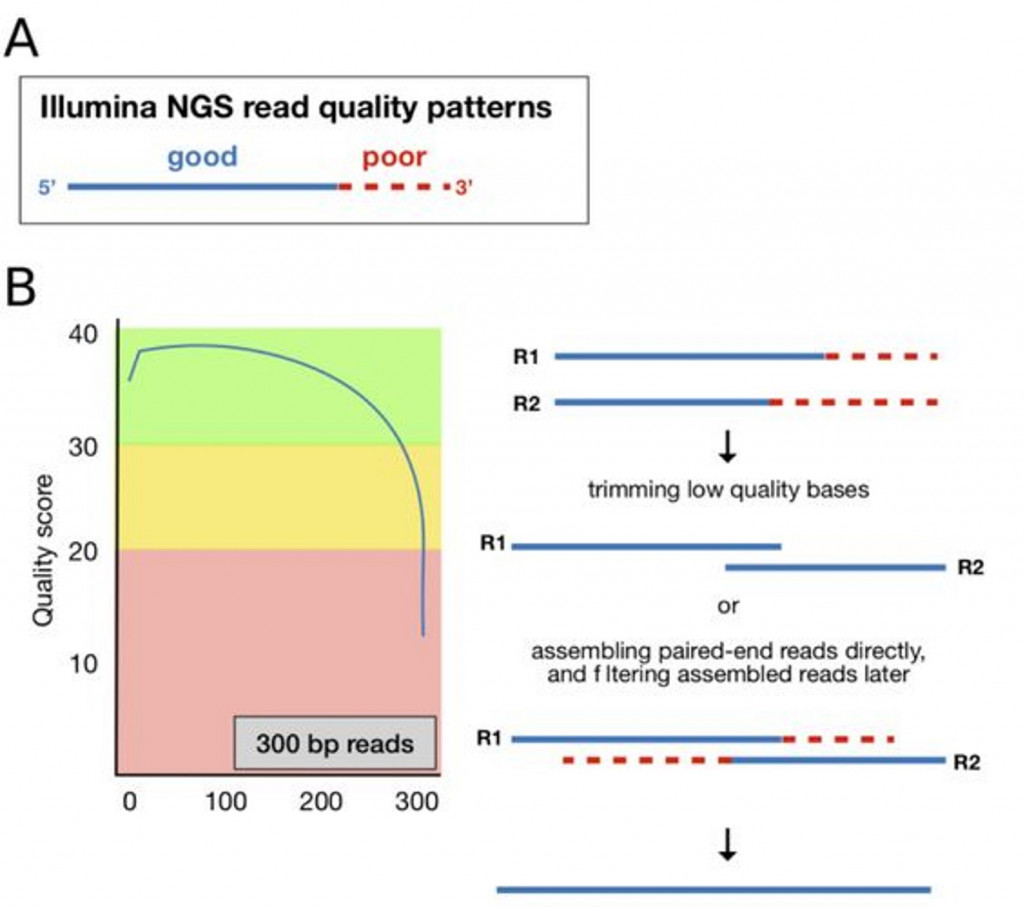
由於定序剛開始與結束的兩端品質都會比較差,
分別從兩端定序的序列也因此補足中間品質差的問題 :
左圖中可見,橫軸為序列長度,縱軸為序列品質分數,越末端品質則越低。在右圖,使用 Pair-end sequencing 可以去除末端品質不佳序列,再利用軟體組裝 (assembling),形成一條 V3-V4 品質佳的序列。
每次提到這裡都覺得人類真的是很聰明。
✏️ 分析
長度問題處理好了,
漸漸的,研究人員擬定了一系列 16S rRNA 鑑種的 Pipeline (分析流程),
除了能知道序列是屬於什麼菌,也想了解群體間的組成、差異(物種多樣性等)。
QIIME2 這套軟體就這樣誕生了,
接下來二十幾天都會講這個部分。
QIIME2 (念起來像Train2) 是一款用於分析微生物菌相的軟體,
輸入端 (Input) 是定序出來的原始檔案 (.fastq),
並輸出 (Output) 各類表格與視覺化檔案 (.tsv, .csv, .qzv, .qza)。
(Bolyen, Evan, et al., 2019)
除了 QIIME2 外,也會搭配一些插件與有趣的工具,
讓整個視覺化之旅變得更豐富~(苦中作樂
在開始安裝前,我們先了解 QIIME2 能夠帶給我們什麼?
1. Denoise (Quality Control, QC)
篩選可信度低的序列,留下品質好的序列。
2. Taxonomy Classification
像是查字典般,利用資料庫與分類器,找出每條序列所對應的菌名(階層)
3. Diversity Analysis
將樣本內、樣本間的菌種們,進行多樣性分析
N. Functional Prediction
後來很多插件工具因應 QIIME2 而生,
可以使用 QIIME2 產出的檔案,進行代謝途徑、酵素預測,
能更清楚這些菌平時在環境中扮演的角色是什麼。
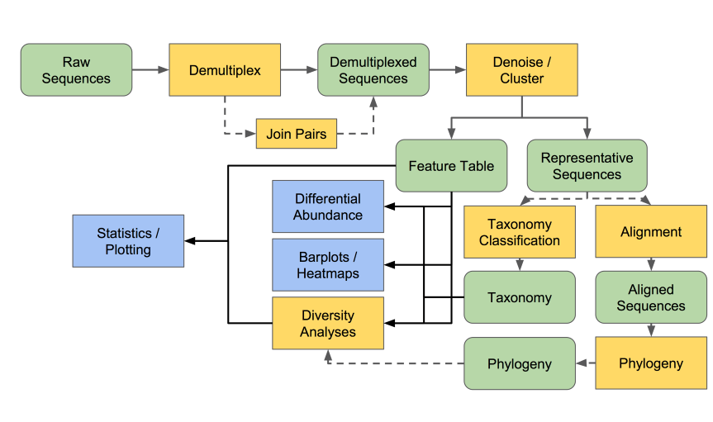
QIIME2 實際上可以做的事情很多,這邊以基礎分析為主
(黃色表示工具;綠色、藍色表示輸入/輸出資料)
本篇文章同步刊載於科學毛怪部落格 PetSci Blog。