QIIME2 對於二代定序資料高度支援,
但在第三代定序的資料相容性上仍未完善,
本篇採用由 PacBio 官方於 2022.8 出品,
結合 QIIME2 等眾多套件的超級方便軟體 pb-16S-nf,
並擁有下列特色 :
仔細看標題軟體版本為 v0.3 難不成是測試版 ?
對,就是拿麼新,你已經站生物資訊的浪頭上了( •̀ ω •́ )✧
這裡附上一張 PacBio HiFi 平台與其他產品/競品比較,
使用自己的序列檔案時要留意是否符合下圖綠色框框的標示 :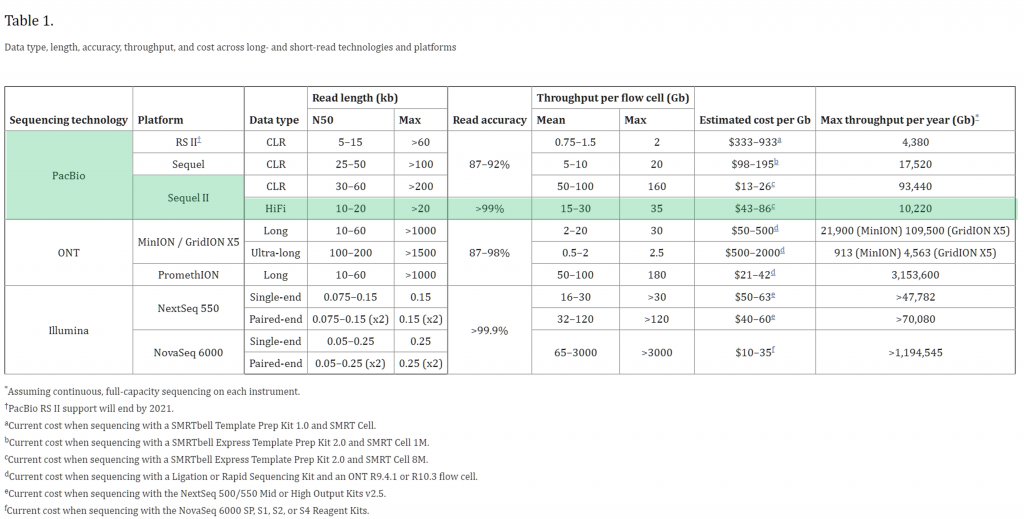
(Logsdon, G. A., Vollger, M. R., & Eichler, E. E., 2020)
根據官方釋出資料,會發現其流程與[Day 05]介紹的 QIIME2 大致一致,
圖中包含 :
Input -> Trimming -> QC -> Taxonomy classification -> Phylogenic tree, diversity analysis, visualization ...
觀察紅色字體部分,可以發現與先前介紹的步驟相當類似。若沒有分析過次世代定序資料,強烈建議先實作完 NGS 分析再學習三代會比較上手 !
Reference : pb-16S-nf
比起 QIIME2 本身, pb-16S-nf 又囊括了更多套件補足 QIIME2 在三代分析的不足,
甚至提供一鍵跑完上述所有流程 !
conda 建置環境與安裝 Mamba 及 Nextflow 輔助套件
conda create --name TGS python=3.8
conda activate TGS
#建立一個名為 TGS 虛擬環境,並進入環境
conda install mamba -n base -c conda-forge
mamba init
#多執行緒版 conda install,若是一路學習過來的讀者,mammba 已在 [Day 19] 安裝,可跳過這兩行
conda install -c bioconda nextflow
#提供自動執行 Pipeline、中斷能繼續分析的軟體,pb-16S-nf 需要它
下載 pb-16S-nf 主程式
git clone https://github.com/PacificBiosciences/pb-16S-nf.git
#這個連結會下載最新版本的pb-16S-nf
#############################
#2023.01.29 補充說明 :
#若在後續步驟中發現不明原因的報錯,
#可以嘗試重新安裝本篇採用的v0.3版本 :
wget https://github.com/PacificBiosciences/pb-16S-nf/archive/refs/tags/v0.3.tar.gz
#下載v0.3版本
tar zxvf v0.3.tar.gz
#解壓縮
mv pb-16S-nf-0.3 pb-16S-nf
#更改檔名之後(以便與後續教學吻合)可前往下一步驟
修正配置檔案 (v0.3版才需要做這步驟,新版可跳下一點:初始化與下載資料庫和分類器)
這裡有個神奇 Bug ,會導致安裝失敗,
需要修正配置檔案 qiime2-2022.2-py38-linux-conda.yml
我們需要先將 bioconductor-genomeinfodbdata 版本要求調整 :
cd pb-16S-nf/env
#進去 pb-16S-nf/env資料夾
vi qiime2-2022.2-py38-linux-conda.yml
#編輯 qiime2-2022.2-py38-linux-conda.yml
找到 bioconductor-genomeinfodbdata=1.2.7,
將其改為 :
bioconductor-genomeinfodbdata=1.2.6
不知道什麼原因 1.2.7 載點失效,於是降版。
:wq 儲存後,回到 pb-16S-nf 資料夾 :
cd ..
初始化與下載資料庫和分類器
過程會有點久,可以先去看場電影,可以開 screen 跑 :
nextflow run main.nf --download_db
#開始下載物種資料庫與分類器
完成畫面 :
executor > Local (1)
[8b/ae67a7] process > pb16S:download_db [100%] 1 of 1 ✔
'Completed at: 04-Oct-2022 00:00:08
Duration : 12m 22s
CPU hours : 0.8
Succeeded : 1'
echo -e "sample-id\tabsolute-filepath\ntest_data\t$(readlink -f test_data/test_1000_reads.fastq.gz)" > test_data/test_sample.tsv
#Create sample TSV for testing
nextflow run main.nf \
--input test_data/test_sample.tsv \
--metadata test_data/test_metadata.tsv \
-profile conda \
--outdir results
#運行小小的測試資料,這指令內容是不是有點熟悉~
順利的話會出現如下圖所示的提示訊息,就可以快樂前往下一天 :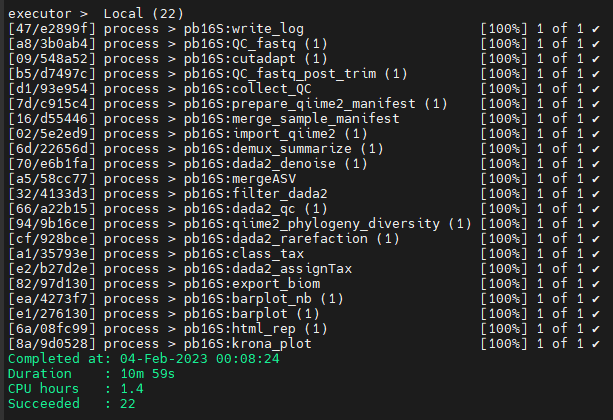
如果有未安裝完全的環境,在過程中會自動補齊,
補齊過程有點久,可以去運動再回來。
Reference : pb-16S-nf
如果遇到紅字報錯且含有以下文字:
Command error:
.command.sh: line 2: csvtk: command not found
執行以下指令補安裝csvtk套件,並刪除先前的測試資料紀錄 :
conda install -c bioconda csvtk
rm -r report_results
再重新執行測試資料分析 :
nextflow run main.nf \
--input test_data/test_sample.tsv \
--metadata test_data/test_metadata.tsv \
-profile conda \
--outdir results
你好,感謝詳細的教學
我按照上述步驟(v0.3)進行安裝,但是出現以下錯誤:
Error executing process > 'pb16S:QC_fastq (1)'
Caused by:
Process pb16S:QC_fastq (1) terminated with an error exit status (255)
Command executed:
seqkit fx2tab -j 8 -q --gc -l -H -n test_1000_reads.fastq.gz | csvtk mutate2 -C '%' -t -n sample -e '"test_data"' > test_data.seqkit.readstats.tsv
seqkit stats -T -j 8 -a test_1000_reads.fastq.gz | csvtk mutate2 -C '%' -t -n sample -e '"test_data"' > test_data.seqkit.summarystats.tsv
seqkit seq -j 8 --min-qual 20 test_1000_reads.fastq.gz --out-file test_data.filterQ20.fastq.gz
echo -e "test_data "$PWD"/test_data.filterQ20.fastq.gz" >> test_data_filtered.tsv
Command exit status:
255
Command output:
(empty)
Command error:
.command.sh: line 2: seqkit: command not found
[ERRO] empty file: -
Work dir:
/home/lab617/YY/CCKuopacbio/pb-16S-nf/work/7b/34139fef1c1ebe32920c2033ade0ec
Tip: you can try to figure out what's wrong by changing to the process work dir and showing the script file named .command.sh
我有試過錯誤中描述的指令看起來沒問題,請問你有遇過類似的錯誤嗎?
看起來是環境沒有裝好,蠻多使用者有回報這類問題,可能目前還是測試版關係。可以確認家目錄有無nf_conda資料夾,內含pb-16S開頭資料夾兩個與qiime2資料夾一個。
如果都沒有或是不完整,建議把整個nf-conda資料夾移除,重新執行文章中測試是否安裝成功那一點的指令,讓它自行重新安裝環境。

