
pb-16S-nf 沿襲了 QIIME2 [Day 06] 的特色,
同樣需要下列輸入檔案,在格式上與次世代相當類似 :
(.fastq.gz or .fastq)
以下範例程式運行會耗時半天以上,使用國網(NCHC)或自家伺服器>32 CPUs較佳,
若時間有限會建議所有定序檔案路經改為pb-16S-nf/test_data/test_1000_reads.fastq.gz,會跑很快~
下載公開文獻資料的原理與安裝請見 [Day 04 ],就不贅述。
本次範例選擇汙水微生物菌相序列作為教學 (LaMartina, E. L., Schmoldt, A. L., & Newton, R. J., 2022) :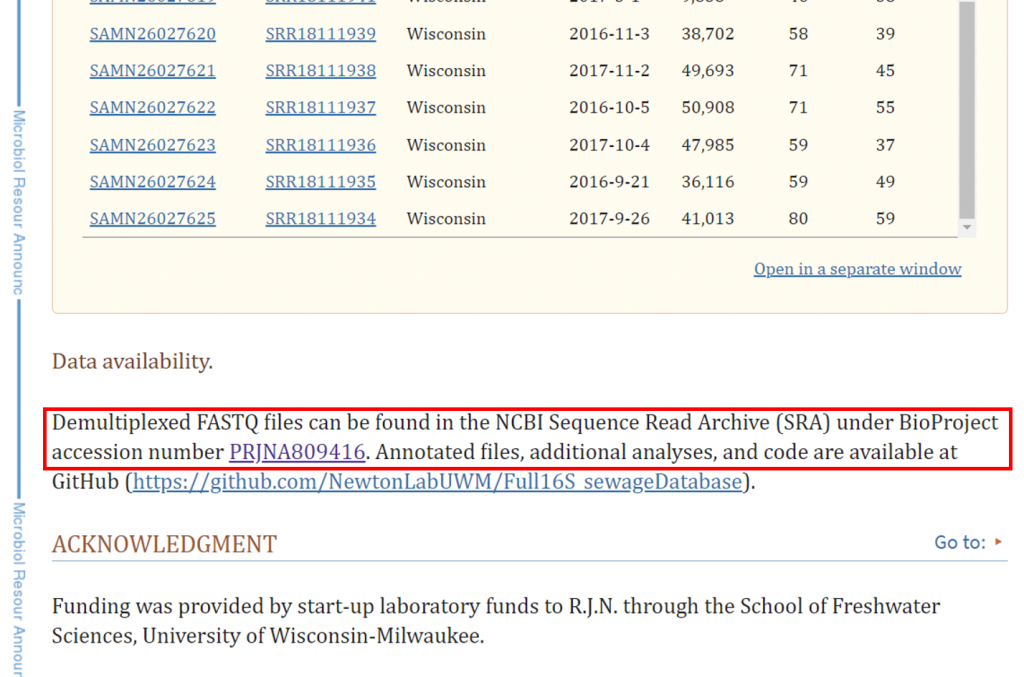
其序列為 PacBio 16S Full Length (27F & 1492R) ,
accession number 為 PRJNA809416 並餵給 SRA Run Selector,
這次選擇四個檔案當示範~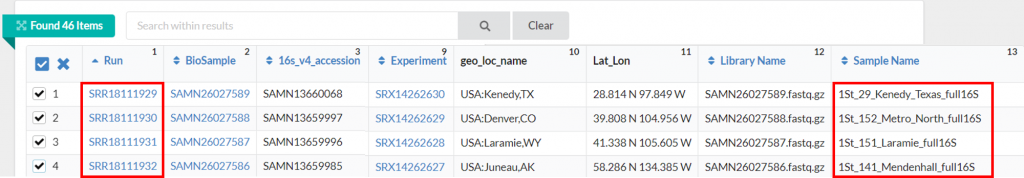
教學方便,我創一個資料夾TGS_sewage在pb-16S-nf下 :
mkdir TGS_oral
cd TGS_oral
將以下序列檔案編號放入 SRR_Acc_List.txt,並放到TGS_sewage下 :
vi SRR_Acc_List.txt
貼上來自 PRJNA809416 的其中四個檔案 :
SRR18111929
SRR18111930
SRR18111931
SRR18111932
製作 batch_download.py :
vi batch_download.py
from bioinfokit.analys import fastq
fastq.sra_bd(file='SRR_Acc_List.txt', t=16, other_opts='--split-files' )
啟動SRA_download虛擬環境並執行 batch_download.py,
這裡會需要一些時間,可以去買杯咖啡~
conda activate SRA_download
python batch_download.py
若出現
No module named 'bioinfokit',回到 [Day 04] 安裝 bioinfokit。
將定序檔案壓縮 :
gzip *.fastq
順利的樣子 :
XXXXXX users 45418123 Oct 4 00:27 SRR18111929.fastq.gz
XXXXXX users 37210512 Oct 4 00:19 SRR18111930.fastq.gz
XXXXXX users 54730305 Oct 4 00:32 SRR18111931.fastq.gz
XXXXXX users 37175973 Oct 4 00:35 SRR18111932.fastq.gz
manifest 是清單的意思,與 QIIME2 輸入格式相同,
檔名為 manifest.tsv,路徑XXXXXX記得更改成自己的
sample-id absolute-filepath
1St_29_Kenedy_Texas /home/XXXXXX/pb-16S-nf/TGS_sewage/SRR18111929.fastq.gz
1St_152_Metro_North /home/XXXXXX/pb-16S-nf/TGS_sewage/SRR18111930.fastq.gz
1St_151_Laramie /home/XXXXXX/pb-16S-nf/TGS_sewage/SRR18111931.fastq.gz
1St_141_Mendenhall /home/XXXXXX/pb-16S-nf/TGS_sewage/SRR18111932.fastq.gz
若時間有限會建議所有定序檔案路經改為
pb-16S-nf/test_data/test_1000_reads.fastq.gz發現了嗎 ?
與 NGS V3-V4 雙尾定序的資料不同,每個樣本僅有一個定序檔案,
因為突破了技術限制,不須雙邊讀取,
metadata 是後設資料、註釋資料的意思,
可以根據實驗設計新增新的 column ,
與次世代分析相同,新增index一個樣本一組,
範例中也新增組別名為group分別為A, B
檔名為 sample-metadata.tsv :
sample_name index group
1St_29_Kenedy_Texas 1St_29_Kenedy_Texas A
1St_152_Metro_North 1St_152_Metro_North A
1St_151_Laramie 1St_151_Laramie B
1St_141_Mendenhall 1St_141_Mendenhall B
