有別於 QIIME2 一步一步手動輸入指令處理,
pb-16S-nf 與 PICRUSt2 一樣提供單一指令執行全部流程,
所需要的檔案就是前面所提的.fastq.qz、manifest.tsv、sample-metadata.tsv :
nextflow run main.nf \
--input TGS_sewage/manifest.tsv \
--metadata TGS_sewage/sample-metadata.tsv \
-profile conda \
--outdir TGS_sewage/allResults \
--dada2_cpu 32 \
--cutadapt_cpu 32 \
--vsearch_cpu 32
--outdir : 輸出的資料夾名稱,
--dada2_cpu, --cutadapt_cpu, --vsearch_cpu : 運行核心數調整。
另外,可以再加上--run_picrust2 進行基因功能分析。
這是最順利的分析完成畫面,
可以看到先前介紹的步驟組成一整條Pipeline,
綠色顯示完成時間、耗時與過程等等,
如果是照著前一天步驟下載SRA序列資料分析,
大約會跑幾個小時,推薦放著去看個影集再回來~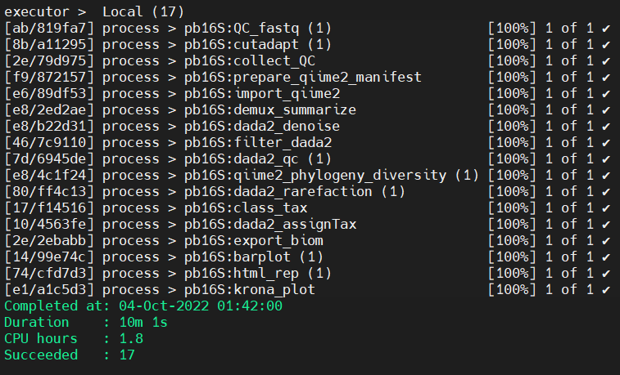
實務上往往遇到報錯情形,如註釋資料或序列檔案的問題等,
pb-16S-nf 優勢在於採用 Nextflow 套件,
幫助報錯修正後仍可用同一指令接續執行,續分析與上述指令相同,
不同的是多加上了一條參數 -resume 於指令中 :
nextflow run main.nf \
--input TGS_sewage/manifest.tsv \
--metadata TGS_sewage/sample-metadata.tsv \
-profile conda \
--outdir TGS_sewage/allResults \
--dada2_cpu 32 \
--cutadapt_cpu 32 \
--vsearch_cpu 32 \
-resume
可以發現多了 cached ,代表先前已分析過,會快速地跳過 :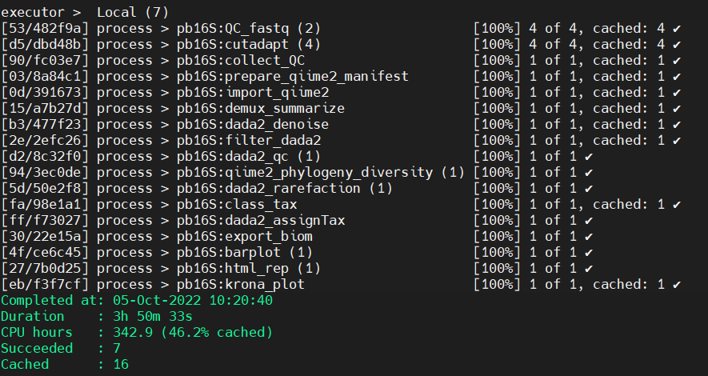
順利的話會得到 allResults 資料夾,裡面包含如下 :
cutadapt_summary
#去除樣本 adapter,概念在 [Day 03] 次世代 PartA 出現
dada2
# QC過後獲得的檔案stats, table, rep-seqs,同 [Day 08]
filtered_input_FASTQ
#樣本 Q score > 20 (預設)的 fastq.qz
import_qiime
# QIIME2 的加工檔案 (.qza),同 [Day 07] 的匯入資料
results
#重要!! 主要分析結果
summary_demux
#樣本拆分的資料,同 [Day 07] 的 demux.qzv 視覺化
trimmed_primers_FASTQ
#樣本剪切掉 primers 的 fastq.qz
位於TGS_sewage/allResults/results/visualize_biom.html,
是一個網頁檔案(.html),拉到本機端直接點開就可以觀看分析的情形,
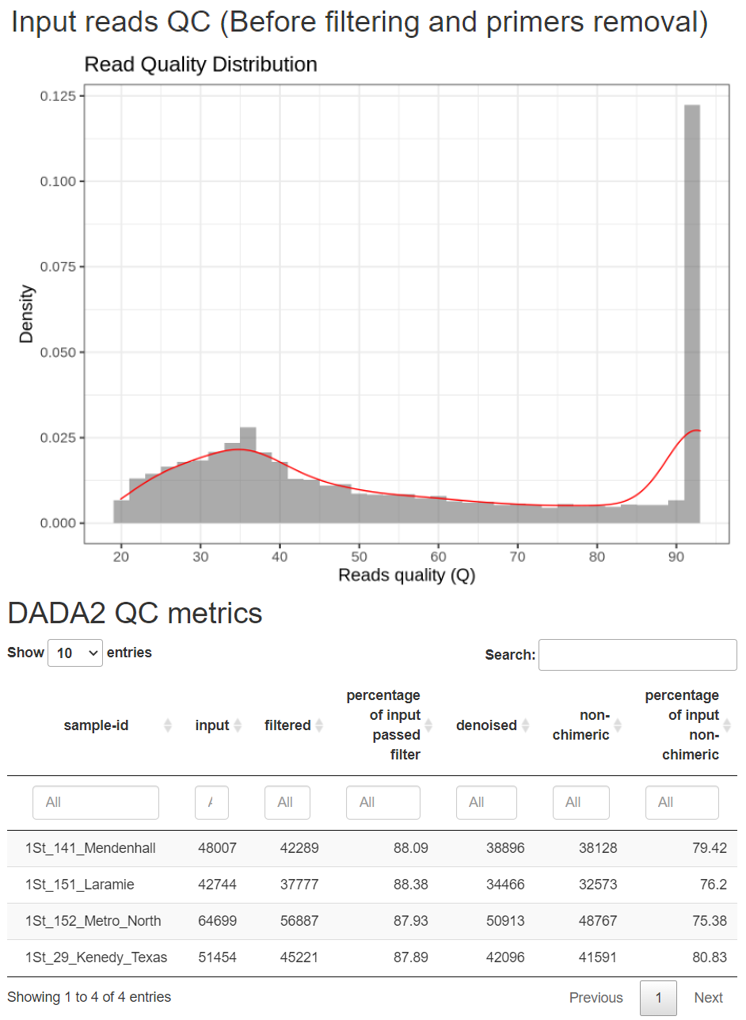
基礎資料包含 QC 的結果、兩種分類器的物種層級狀態等 :
由於省去了手動 Trimming 過程,
改由程式自動判別,基本上 QC 後剩餘的條數都有七成以上 :
因為裡頭太多.qzv、.tsv、.html 可直接檢視的檔案了,為求方便通常會將所有資料夾壓縮打包 :
tar -zcvphf TGS_sewage.tar.gz results
完成後會看到一個可可愛愛的檔案出現,再拉到本機端檢視 :
-rw-r--r-- 1 XXXXXX users 416939627 Oct 6 19:46 TGS_sewage.tar.gz

