
上一篇我們的基因體時代-AI, Data和生物資訊 Day20-註釋基因資訊的BED檔案格式和bedtools上一篇主要先介紹一下BED檔案格式和bedtools,因為這算是整個基因註釋很重要的格式,也因此理解其巨大的可擴展性,只要所有的東西以基因序列的位置作為共同對照,即能整合這些資訊在一起,所以每幾年有新的檢驗技術來量測DNA上的變化時,就會有新的資訊可以形成track來做註釋。
這邊再往下分享這些BED檔案在R裡面怎麼處理,相對於BED檔案可以直接用字串處理工具如awk, sed等方法處理,但實際上也可以利用R語言,相對於使用bedtools在命令行環境下直接單純做處理,在R裡面它的操作可以更為複雜和精緻,在R中處理,主要是利用一個Bioconductor專案中的軟體包,用這些軟體包來幫助處理相關之問題。目前Bioconductor是一個含有936以上個R語言packeges的開源軟體平台,於2001年開始這個專案,其原始開發團隊由加拿大生物統計學家Robert Gentlemen領軍,由美國的Fred Hutchinson Cancer Research center領頭發展,成立的目的是為了提供高通量基因體學研究資料更好的分析工具,相對於R的發展開源且沒有任何資金贊助,bioconductor其實是一個有接受NIH資助的計畫,目的便是發展直接處理高通量資料的工具。
理解所謂的BED檔案格式後,就會發現這其實就是單純用資料表達一段區域,在Bioconductor中則有兩個包用來建構在R裡面處理這類資料。
基因組學的知識本質上就是由一段段線性序列分佈在不同染色體上所建立的,這樣的知識或稱作註釋可以分為兩類,一類是所謂的基因模型(哪邊是外顯子、哪邊是內顯子,哪個區域決定某個最終蛋白質)、轉錄因子的結合位置、特定區域的GC比例、區域的多型性,這類資訊可由各式各樣的生物資料庫所取得,另一類資訊則是實驗室的量測結果如定序資料的等等,為了建議一個可以在R中處理這大類基因註釋資料,Bioconductor團隊裡面的科學家便提出了Ranges的架構。
這個Ranges的資料可以有下面的一些操作: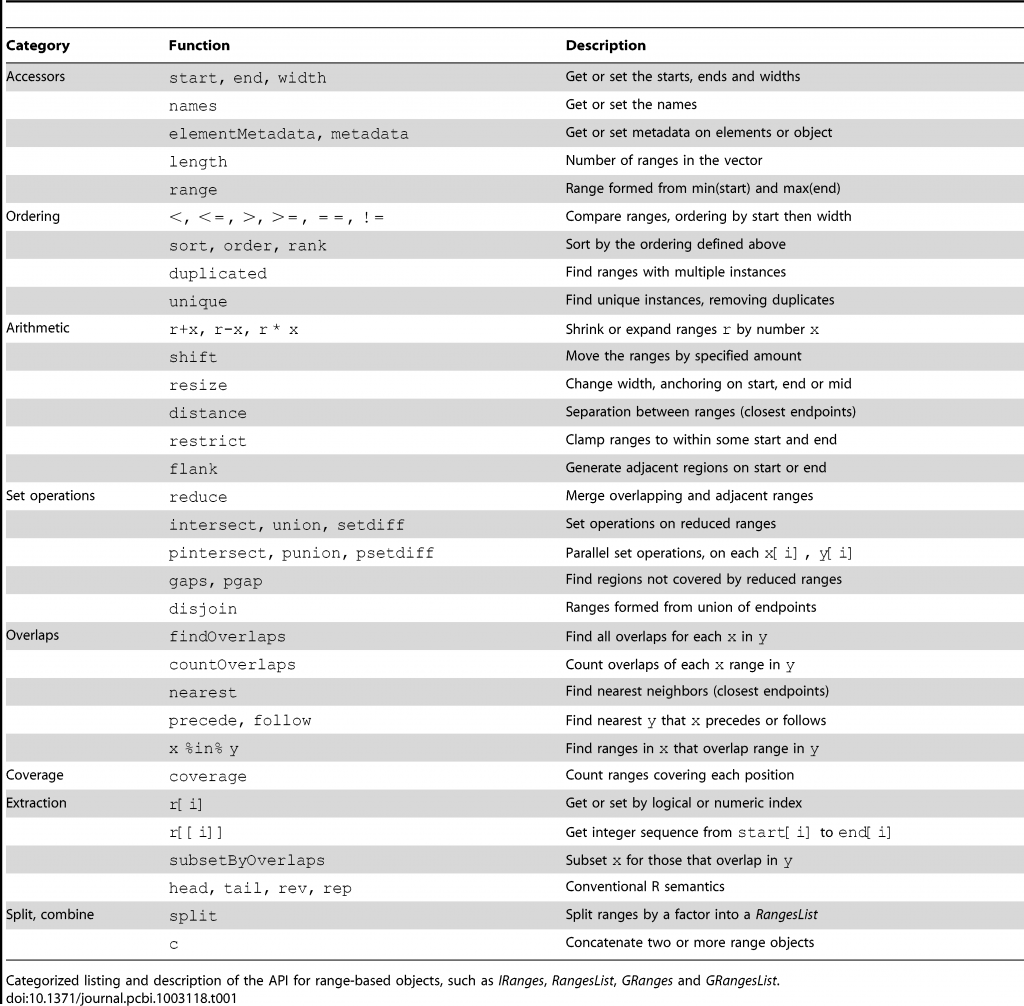
下面的示意圖可以看到在最基礎的Ranges架構中有的計算邏輯,最後建立了這個IRanges和GenomicRanges函數(這邊可以發現跟bedtools部份功能其實是一樣的),你可以去計算兩串區域資料間的一些關係,藉此去進行篩選、合併、取值等等功能。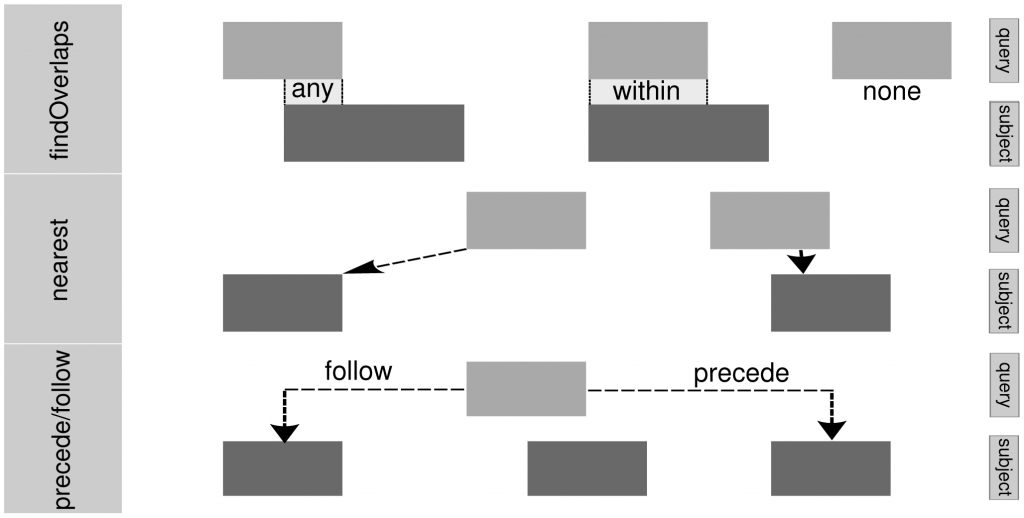
這邊稍微來介紹一下,這邊需要在R環境中先安裝IRanges和GenomicRanges兩個封包才能執行相關的函數。
在R環境中執行,因為這兩個封包是在Bioconductor下面的,相對於一般R的library,使用install.packages來安裝,要安裝Bioconductor下面的library,則必須使用Bioconductor的安裝工具,像在邊就是使用BiocManage封包來進行安裝。
BiocManager::install(c("IRanges", "GenomicRanges"))
安裝完後,在讀取到環境中即可
library(IRanges)
library(GenomicRanges)
一個IRanges物件可以藉由給予四個參數:start, end, width, names就能完成,但最少只要給予start和width即可,兩個的數列必須要等長的,如同下面這樣。在IRanges物件中不包含所在染色體資訊、正負股、或其他複雜註解資訊。
IRanges物件
Gene_range <- IRanges::IRanges(start=c(1,3,5,8,9), width=c(1,2,2,2,2))
將其顯示出來就會長這樣
> Gene_range
IRanges object with 5 ranges and 0 metadata columns:
start end width
<integer> <integer> <integer>
[1] 1 1 1
[2] 3 4 2
[3] 5 6 2
[4] 8 9 2
[5] 9 10 2
GenomicRanges則是在IRanges物件上賦予生物意義,加上每個區域所在之染色體位置、正負股、其他註解,在GenomicRanges物件上的操作,會跟IRanges一樣,這也是library開發者為了讓使用者體驗有一致性所加上的考量。
GenomicRanges物件建立主要需要有三個參數seqnames,ranges,strand,seqnames和strand給予其一個Rle物件,一個是用來給染色體資訊,一個則是用來定義正負股,ranges給予其一個IRanges物件,這個參數的vector長度都要一樣。
genomic_ranges <- GRanges(
seqnames = Rle(c("chr1", "chr2", "chr1", "chr3"), c(1, 3, 2, 4)),
ranges = IRanges(101:110, end = 111:120, names = head(letters, 10)),
strand = Rle(strand(c("-", "+", "*", "+", "-")), c(1, 2, 2, 3, 2)))
將其顯示出來會像這樣
> genomic_ranges
GRanges object with 10 ranges and 0 metadata columns:
seqnames ranges strand
<Rle> <IRanges> <Rle>
a chr1 101-111 -
b chr2 102-112 +
c chr2 103-113 +
d chr2 104-114 *
e chr1 105-115 *
f chr1 106-116 +
g chr3 107-117 +
h chr3 108-118 +
i chr3 109-119 -
j chr3 110-120 -
-------
seqinfo: 3 sequences from an unspecified genome; no seqlengths
但GRanges最重要的特性是可以無限往後面延伸資訊,這邊就讓人想到BED檔案或是VCF檔案格式,比如下面這個就是給每一個Ranges加入統計分數,和GC比例之資訊。
genomic_ranges <- GRanges(
seqnames = Rle(c("chr1", "chr2", "chr1", "chr3"), c(1, 3, 2, 4)),
ranges = IRanges(101:110, end = 111:120, names = head(letters, 10)),
strand = Rle(strand(c("-", "+", "*", "+", "-")), c(1, 2, 2, 3, 2)),
score = 1:10,
GC = seq(1, 0, length=10))
將其顯示出來會像這樣
> genomic_ranges
GRanges object with 10 ranges and 2 metadata columns:
seqnames ranges strand | score GC
<Rle> <IRanges> <Rle> | <integer> <numeric>
a chr1 101-111 - | 1 1.000000
b chr2 102-112 + | 2 0.888889
c chr2 103-113 + | 3 0.777778
d chr2 104-114 * | 4 0.666667
e chr1 105-115 * | 5 0.555556
f chr1 106-116 + | 6 0.444444
g chr3 107-117 + | 7 0.333333
h chr3 108-118 + | 8 0.222222
i chr3 109-119 - | 9 0.111111
j chr3 110-120 - | 10 0.000000
-------
seqinfo: 3 sequences from an unspecified genome; no seqlengths
參考閱讀:
Software for Computing and Annotating Genomic Ranges, 2013, PLoS Computational Biology
我們的基因體時代-Bioconductor:基因體學研究的好幫手(一)
我們的基因體時代-理解Bioconductor系列(一):Bioconductor中的Annotation資源
An Introduction to the GenomicRanges Package. 2021. Bioconductor
Lee, S., Cook, D. & Lawrence, M. plyranges: a grammar of genomic data transformation. Genome Biol 20, 4 (2019). https://doi.org/10.1186/s13059-018-1597-8
Herve Pages, Michael Lawrence. A quick introduction to GRanges and GRangesList object. 2015
Herve Pages. 10 things(maybe) you didn't know about GenomicRanges, Biostrings, and Rsamtools. 2016
trackViewer Vignette: Change the track styles
Florian Hahne, Robert Ivanek. The Gviz User Guide.2021
Common Bioconductor Methods and Classes
Soroor Hediyeh-Zadeh, An introduction to Bioconductor and it's data structure
這個月的規劃貼在這篇文章中我們的基因體時代-AI, Data和生物資訊 Overview,也會持續調整!我們的基因體時代是我經營的部落格,如有對於生物資訊、檢驗醫學、資料視覺化、R語言有興趣的話,可以來交流交流!

 iThome鐵人賽
iThome鐵人賽