
上一篇我們的基因體時代-AI, Data和生物資訊 Day06-蛋白質結構和機器學習01我們繼續分享另一個重要的生物問題,如何從蛋白質序列來預測蛋白質的折疊功能,而這個問題在今年有突破性的進展,將會提速近年來逐漸火熱的合成生物學領域,也就是我們可以用來設計新的蛋白質功能,今年這兩個框架分別是AlphaFold2和 RoseTTAFold,可能會改變整個生物學領域。
這兩個方式分別是Google的DeepMind和David Baker團隊所發展的,只不過相對於DeepMind來說,David Baker算是computation protein design領域的資深研究學家,但就沒有那麼高的名氣,非常推薦他這部Youtube的演講,分享他是怎麼設計針對COVID棘蛋白的小蛋白質(QB3 Webinar: Designing Proteins from Scratch for 21st Century Challenges)
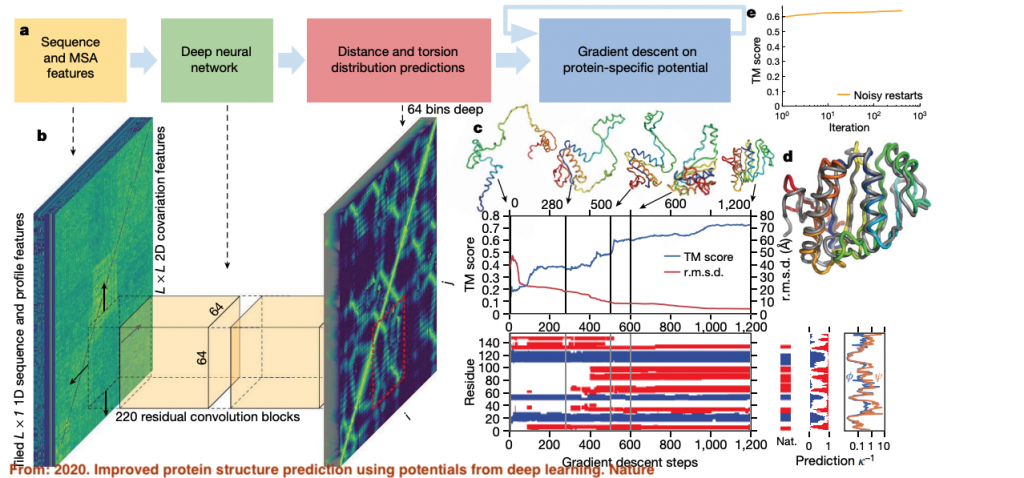
這是AlphaFold1的架構圖,可以跟等等要介紹的AlphaFold2的架構稍微比較一下,從前一篇可以知道,在看這類為了解決生物問題建立的模型,通常最重要的是去釐清他的輸入到底有什麼,這樣本質上會決定可以推演的東西有多少。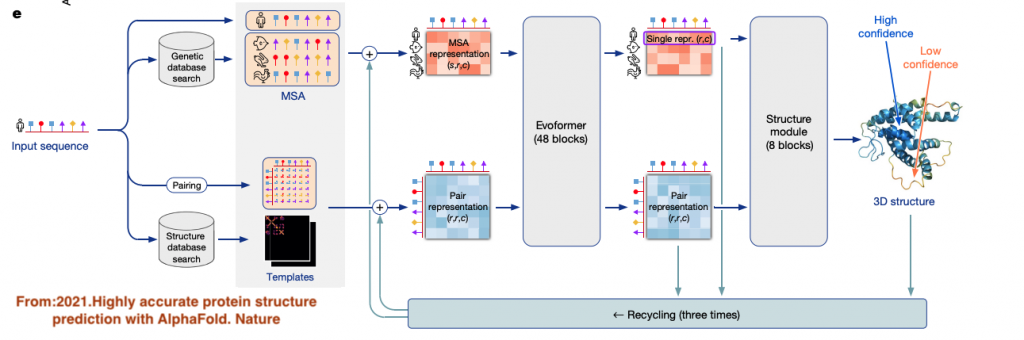
從AlphaFold2的架構,基本上,你會輸入一個想要預測它怎麼折疊的蛋白質序列進去,實際上,整個算法會再去抓取兩個相關資料庫的資料來整合成模型的輸入,第一個就是去看這個序列跟已知的序列資料庫中的哪些蛋白質相似,以及在其他物種中,同樣蛋白質名稱的序列長怎麼樣,將這些不同物種中產生同樣蛋白質功能的序列整合成所謂的MSA(Multiple Sequence Alignment)的資料,長得就像是這樣:
就是把這些類似或同樣功能之蛋白質在不同物種中的序列轉換成像這樣矩陣,就是所謂的MSA,可以看出這在AlphaFold1中也是會用到這個矩陣資料,另外一個資料就是從目前已知的蛋白質結構資料庫中去找尋上面那些MSA中,是否已經有部分結構的資料,將這兩個資訊往下送去兩個模組,一個是Evoformer和Structure module,總共Backward三次。
Evoformer細節的架構如下圖: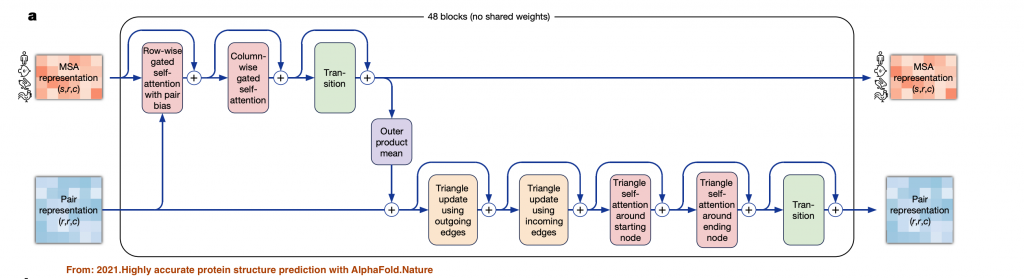
可以看到這邊引入了Attention Models,這邊算是AlphaFold2跟AlphFold1最不一樣的差異,這邊其實是把蛋白質結構預測問題轉換成圖論裡面的關聯性預測,進一步能代表氨基酸之間的距離。
Structure module細節則是如下: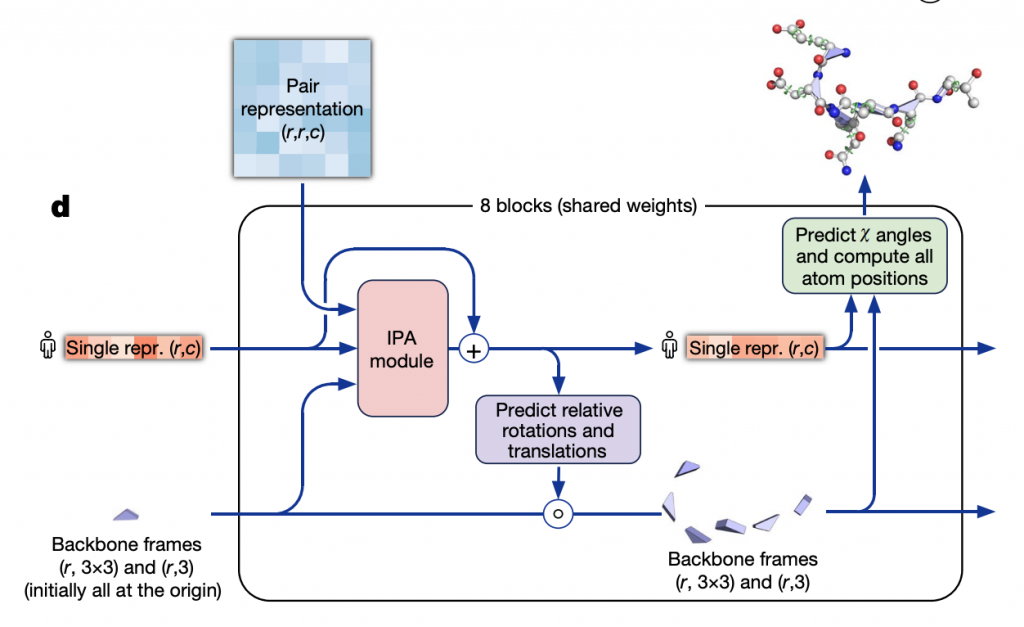
這邊的著重的則是氨基酸序列間的旋轉角度,這邊他們設計了一種Attention Model叫做invariant point attention來處理這個資訊的傳遞和學習。
他們的技術細節可以直接看這個,寫得非常細!
RoseTTAFold的架構可以參考他們發表在Science前上傳到bioRxiv的論文,從文章的開頭可以看出他們算是在AlphaFold2提出後,努力改良提出的版本,所以最大的特色就是可以用較低階的顯卡就在本機做預測:
DeepMind presented remarkably accurate protein structure predictions at the CASP14 conference. We explored network architectures incorporating related ideas and obtained the best performance with a 3-track network in which information at the 1D sequence level, the 2D distance map level, and the 3D coordinate level is successively transformed and integrated.
他們模型建構方式其實就是整合了Domain knowledge的想法(畢竟David Baker多年深厚的功力呀),相對於AlphaFold2的模型架構,這個模型可能在CASP14的Benchmark中表現無法如AlphaFold2一樣,但其對於一些功能性的預測能力較好,下面是他的模型架構: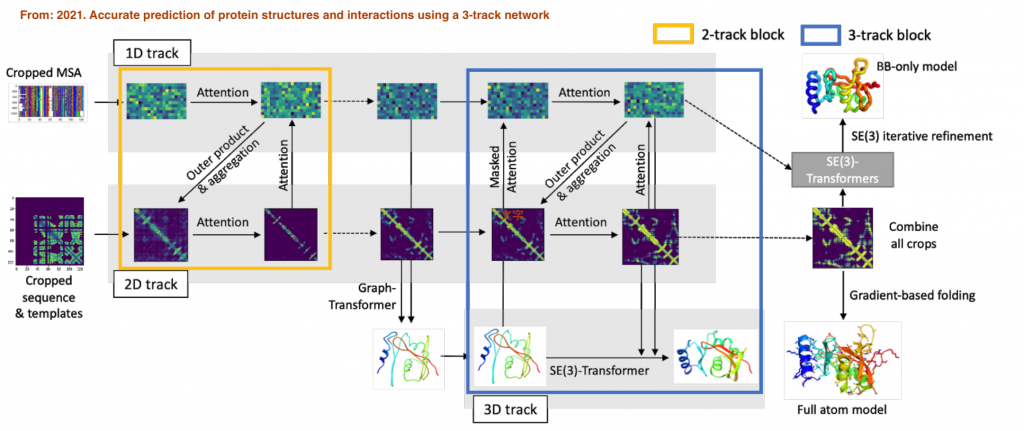
模型的架構也是採用Attention model,但是簡化很多,比較不像DeepMind有很多高級技巧,他們的基本架構是分三個部分來學習,第一個部分是學習蛋白質的一微結構(就是序列),第二部分則是二級結構(alpha helix, beta sheet),最後則是三級結構(就是綜合所有二級結構間的關聯),他們有建立一個可以線上使用他們模型來做分析的,位置在這邊。
閱讀參考:
AlphaFold 2
Senior, A.W., Evans, R., Jumper, J. et al. Improved protein structure prediction using potentials from deep learning. Nature 577, 706–710 (2020). https://doi.org/10.1038/s41586-019-1923-7
RoseTTAFold
Accurate Prediction of protein structures and interactions using a three-track neural netwrok. 2021. Science
這個月的規劃貼在這篇文章中我們的基因體時代-AI, Data和生物資訊 Overview,也會持續調整!我們的基因體時代是我經營的部落格,如有對於生物資訊、檢驗醫學、資料視覺化、R語言有興趣的話,可以來交流交流!

 iThome鐵人賽
iThome鐵人賽