上一篇我們的基因體時代-AI, Data和生物資訊 Day14- 第二代定序(次世代定序)和它的資料格式上一篇介紹次世代定序之後的檔案格式fastq和相關的軟體和分析工具,接者再往下來到越來越複雜的序列拼裝後的檔案格式:bam和sam,這可能也要先介紹一下背景知識。
從上一篇我們知道從第二代定序機器出來的檔案fastq中,可能會存放上百萬個序列,是從定序機器得到的資料,但這些片段是來自於哪一個染色體或是位置,是不知道的,必須用演算法去做對照,有點像是拼拼圖一樣,如同下圖: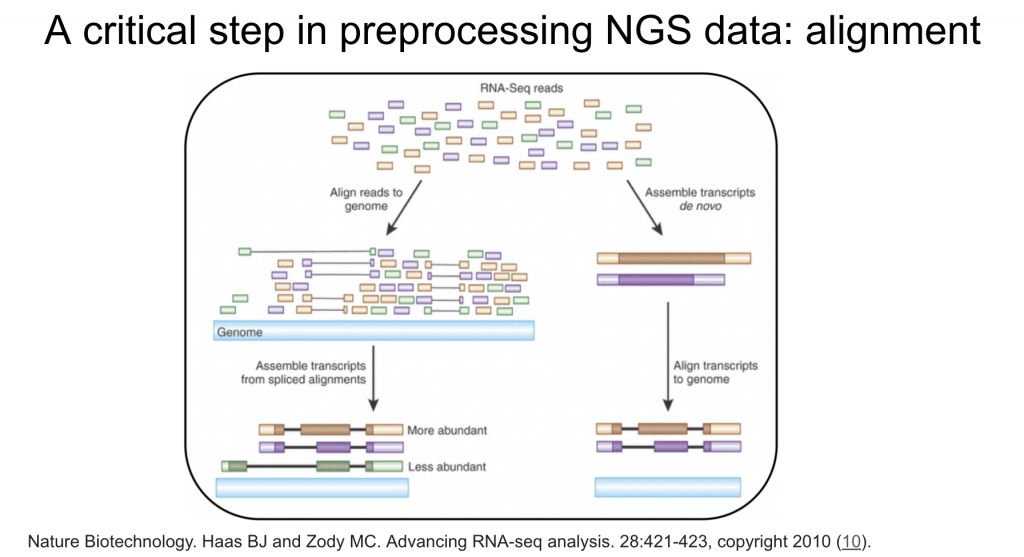
所以辨認每一個定序出來的片段來自何方的時候,我們必須要有個參照序列,就是所謂的參考基因組,但這個參考基因組跟實際上的定序對象的序列是有差異的(人跟人都會有基因序列的差異,所以用的參考基因組也會跟片段有所差異,所以這個比對是有一定的匡鬆條件,才能找到那些個人的變異資訊)。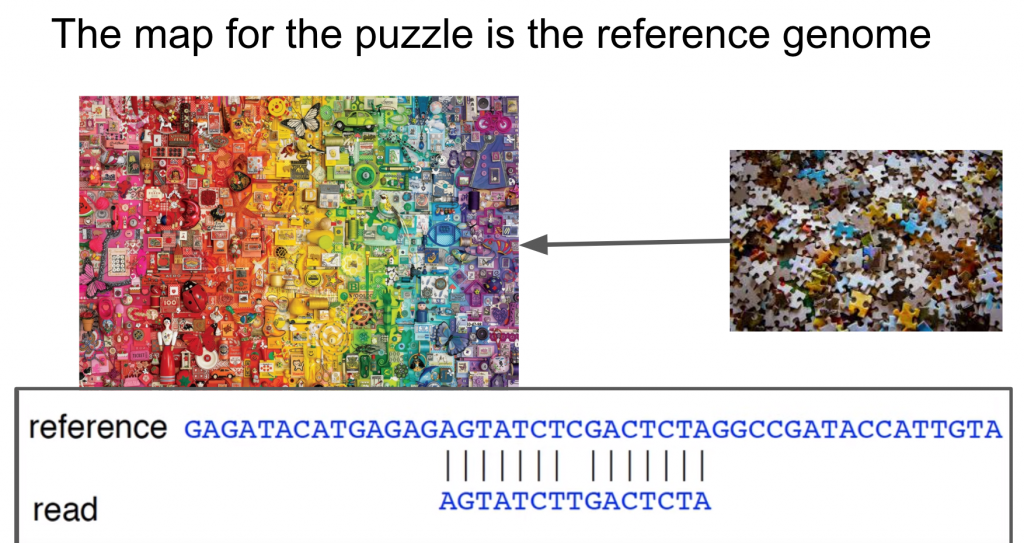
所以當我們把這些片段比對到參考基因組上時,就必須要有另一個格式來儲存這樣的資料。
SAM這個格式就是為了要把比對參考序列的資料儲存起來,而BAM檔就是其二進位的格式,因為假如所有資料都是以text檔案格式存放的話,他實際的檔案大小是非常大的。提到SAM檔案格式就要提到一個領域大神Heng Lin,他撰寫的演算法和軟體基本上是目前所有第二代定序分析軟體的基石,像是bwa, samtools, 在2009年這篇論文中,Heng Lin提出的SAM檔案格式也算是解決領域內資料統合的問題(有點像是fastq或是fasta格式的提出,也是為了統一當時的生物資料交流格式)。
詳細的SAM格式,可以看這份最新的spec文件。可以看SAM格式說明書中的簡易範例
@HD VN:1.6 SO:coordinate
@SQ SN:ref LN:45
r001 99 ref 7 30 8M2I4M1D3M = 37 39 TTAGATAAAGGATACTG *
r002 0 ref 9 30 3S6M1P1I4M * 0 0 AAAAGATAAGGATA *
r003 0 ref 9 30 5S6M * 0 0 GCCTAAGCTAA * SA:Z:ref,29,-,6H5M,17,0;
r004 0 ref 16 30 6M14N5M * 0 0 ATAGCTTCAGC *
r003 2064 ref 29 17 6H5M * 0 0 TAGGC * SA:Z:ref,9,+,5S6M,30,1;
r001 147 ref 37 30 9M = 7 -39 CAGCGGCAT * NM:i:1
簡單來說,SAM是一個tabl分隔的文字資料格式,其分成兩個部分:
Header的部分,每一行是一筆資訊,且是以@開頭,裡面有儲存整個比對中參考的染色體,@後面會有兩個文字作為header資訊種類,至少有@HD(基本文件說明), @SQ(參考序列), @RG(片段分組), @PG(所用的比對分析程式和所下參數), @CO(註解)。



反之,在Alignment的部分則是以tab相隔的格式,裡面共有11欄,每一欄的內容如下:
第一欄QNAME:代表此片段的獨立名稱,第二欄FLAG:為獨立編碼來代表此片段的比對狀況,詳細數字對應其狀況,可由下面的圖表來理解 ,第三欄RNAME:代表此片段比對到的參考基因組其名字為何,通常就是對應@SQ的名稱,第四欄POS:代表所對應到參考基因組的起始位置(從1開始的索引系統1-based system),第五欄MAPQ:代表期比對品質,數字越小代表此片段比對錯誤的機率越大,第六欄CIGAR:代表此片段跟參考序列之間的關係為何,也是有一個表可以對照
,第三欄RNAME:代表此片段比對到的參考基因組其名字為何,通常就是對應@SQ的名稱,第四欄POS:代表所對應到參考基因組的起始位置(從1開始的索引系統1-based system),第五欄MAPQ:代表期比對品質,數字越小代表此片段比對錯誤的機率越大,第六欄CIGAR:代表此片段跟參考序列之間的關係為何,也是有一個表可以對照 ,第七欄RNEXT:代表這個片段的另一組其所比對到的參考基因組名稱(這跟定序方法有關,有所謂的paired-reads,此時也是@SQ的名稱為止),第八欄PNEXT:則是此片段的另一組片段其比對到的位置(1-based system),第九欄TLEN:所在正股還是負股,第十欄SEQ:序列本身,第十一欄QUAL:品質資料。
,第七欄RNEXT:代表這個片段的另一組其所比對到的參考基因組名稱(這跟定序方法有關,有所謂的paired-reads,此時也是@SQ的名稱為止),第八欄PNEXT:則是此片段的另一組片段其比對到的位置(1-based system),第九欄TLEN:所在正股還是負股,第十欄SEQ:序列本身,第十一欄QUAL:品質資料。
理解這個資料格式的內容,那麼接下來就可以分享用來處理和分析相關資料的工具!
相關參考閱讀:
我們的基因體時代:samtools 使用
我們的基因體時代:SAM, BAM and CRAM
Sequence Alignment/Map Format Specification
Li H, Handsaker B, Wysoker A, Fennell T, Ruan J, Homer N, Marth G, Abecasis G, Durbin R; 1000 Genome Project Data Processing Subgroup. The Sequence Alignment/Map format and SAMtools. Bioinformatics. 2009 Aug 15;25(16):2078-9. doi: 10.1093/bioinformatics/btp352. Epub 2009 Jun 8. PMID: 19505943; PMCID: PMC2723002.
Danecek P, Bonfield JK, Liddle J, Marshall J, Ohan V, Pollard MO, Whitwham A, Keane T, McCarthy SA, Davies RM, Li H, Twelve years of SAMtools and BCFtools, GigaScience (2021) 10(2) giab008
這個月的規劃貼在這篇文章中我們的基因體時代-AI, Data和生物資訊 Overview,也會持續調整!我們的基因體時代是我經營的部落格,如有對於生物資訊、檢驗醫學、資料視覺化、R語言有興趣的話,可以來交流交流!


 iThome鐵人賽
iThome鐵人賽