上一篇我們的基因體時代-AI, Data和生物資訊 Day13- 最基本的生物資訊資料格式Fasta 昨天稍微調整步伐,怕原本規劃的第一部份談論太久,所以開始進到當初規劃的路線,上一篇就從生物資訊裡面最基礎的資料格式Fasta開始分享,這個資料格式可以用來儲存DNA序列或是氨基酸序列,且裡面可以編碼從什麼資料庫而來。
那這類的fasta格式在一代定序時代是非常足夠使用的,但直到人類基因體計畫的關係,開始發展能一次定序許多片段的技術,俗成的第二代定序,這時候便開始須要擴增原本的資料格式已便能納進去新技術的資訊,這個格式的名稱叫做fastq,原本其實算是科學家自己用來做紀錄的一種格式,並沒有正式的公開,據說一開始是由Wellcome Trust Sanger Institute的生物學家Jim Mullikin所開始使用的,後來在2004年,由次世代定序公司Solexa所發表,後來這間公司被現在市場最大份額的次世代定序公司Illumina所併購,慢慢的這個格式開始普及。
次世代定序的重點在於一次定序能定序幾百萬個基因片段,每個片段介於150上下,但其中的方法學可以有很多種變形,目前市佔率最高的是illumina這間公司的次世代定序平台,他定序的流程大概如下圖所示: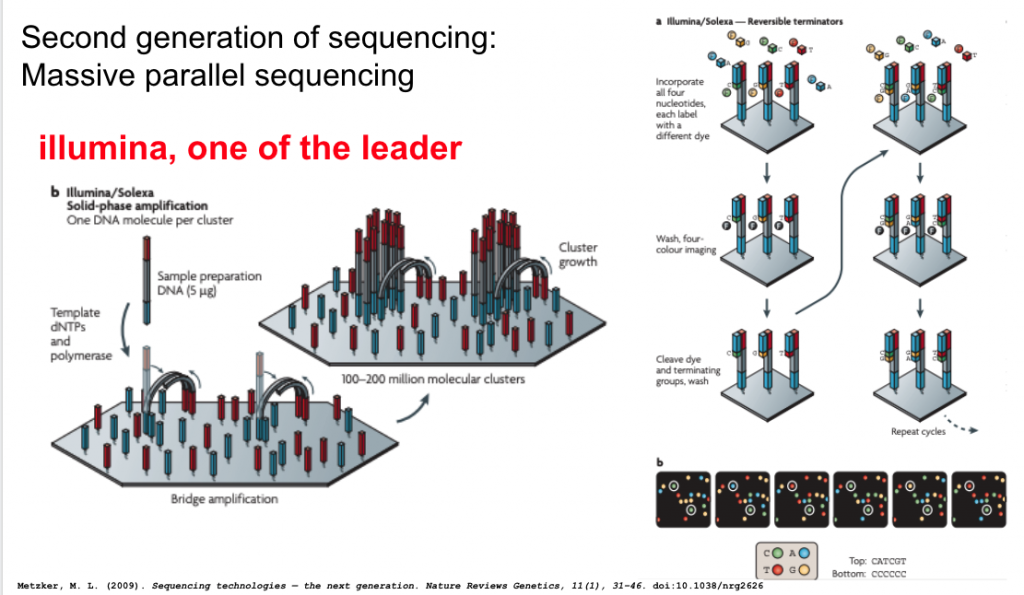
簡化來說,他可以在一個晶片上同時定序數百萬的片段,方法是將經過處理之待定序的片段,黏到晶片上面,會先將這個待定序片段在原地複製成一叢後,進一步利用螢光分子標定的方式,開始定序的時候,其實就是將螢光標定的鹼基材料方進去,它一複製就會放出他對訂的螢光,同時用高速照相機捕捉,在轉換成定序資料,這些序列資料除了當下這個位置的序列外,還會有一個品質數值,就是這個位置螢光的純度,比如這個晶片位置上的點100%都是紅光,那代表這個位置的可信度高,要是這個位置四種螢光都有偵測到,那麼就是不太可信,因此這類的定序方式每個定序資料都有相對應的品質數值。
下面便是一個示範的fastq一筆資料的樣子
@SEQ_ID
GATTTGGGGTTCAAAGCAGTATCGATCAAATAGTAAATCCATTTGTTCAACTCACAGTTT
+
!''*((((***+))%%%++)(%%%%).1***-+*''))**55CCF>>>>>>CCCCCCC65
在fastq中一筆資料基本上有四行組成:
從fastq開始,便進入了需要各式各樣軟體和算法的世界,這邊就可以分成在三個環境中所使用的工具,分別是在linux環境、R及python,這也是基本進入生物資訊領域需要會的技能點數。通常fastq的檔案是當檢體中核酸送進去次世代定序機台後,出來的原始資料,也可以把它當作是:將生物密碼數位化的第一個媒介(好像有點太狗血的形容XD)
大部分生物資訊牽涉到次世代定序資料的處理,很多都是Linux環境中的軟體,基本上,老牌軟體FastQC是大家入門都會使用到的軟體,他可以輸出一份關於這個fastq檔案的品質分析資料,如下面的資訊: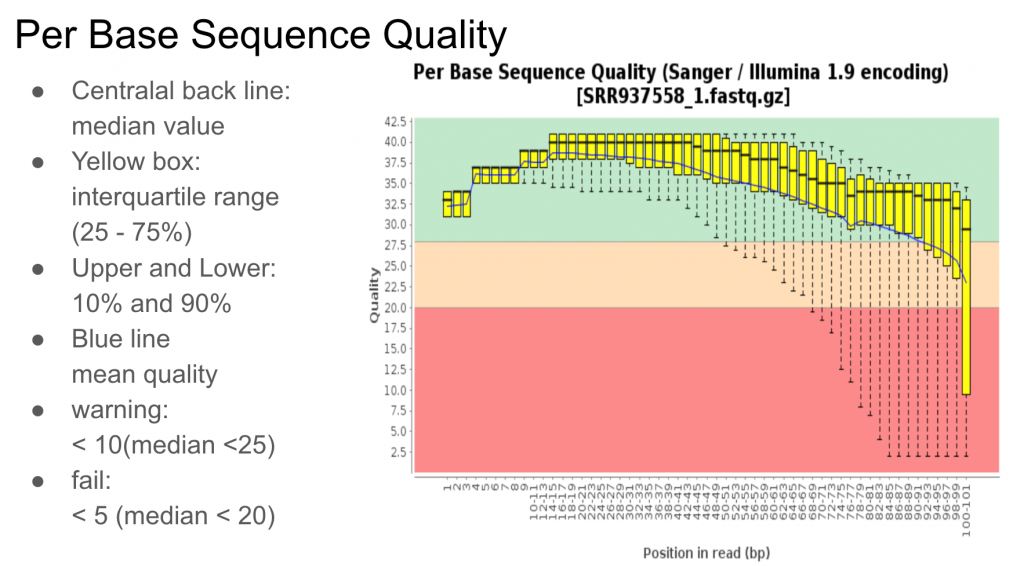
會顯示出每個定序資料長度排在一起,每一個位置其平均的品質條件如何的資訊,根據這個資訊可以用來判斷是否要來做篩選或是剔除前或是後一些品質比較差的序列。
安裝後,直接在命令行執行下面的參數就可以,它會自動偵測你指定的資料夾中的fastq檔案,並且做分析。
fastqc seqfile1 seqfile2 .... seqfileN -o outputfile
再來當我們想要去篩選或是清理這些定序出來的資料,把特定序列清除、不好品質的序列刪掉,那麼就可以使用fastp款軟體,以前的話,可能要用多個軟件,比如fastqc+trimmomatic,但現在fastp一個就可以完成,且速度也蠻快的,另外,它輸出的報告也頗美觀.
除此之外,seqtk和seqkit也都是Linux環境下可用的工具。
Bioconductor是R裡面一個針對生物資訊所創立的大Project,裡面有幾乎所有你需要用到做生物資訊分析的library,在針對fastq檔案的處理上,可以使用ShortRead, Biostrings, qrqc來處理
在python中則有Biophython可以來處理。
Chen S, Zhou Y, Chen Y, Gu J. fastp: an ultra-fast all-in-one FASTQ preprocessor. Bioinformatics. 2018 Sep 1;34(17):i884-i890. doi: 10.1093/bioinformatics/bty560. PMID: 30423086; PMCID: PMC6129281.
Philip Ewels, Måns Magnusson, Sverker Lundin, Max Käller, MultiQC: summarize analysis results for multiple tools and samples in a single report, Bioinformatics, Volume 32, Issue 19, 1 October 2016, Pages 3047–3048, https://doi.org/10.1093/bioinformatics/btw354
Metzker, M. L. (2009). Sequencing technologies — the next generation. Nature Reviews Genetics, 11(1), 31–46. doi:10.1038/nrg2626
Cock PJ, Fields CJ, Goto N, Heuer ML, Rice PM. The Sanger FASTQ file format for sequences with quality scores, and the Solexa/Illumina FASTQ variants. Nucleic Acids Res. 2010;38(6):1767-1771. doi:10.1093/nar/gkp1137
Goodwin, S., McPherson, J. & McCombie, W. Coming of age: ten years of next-generation sequencing technologies. Nat Rev Genet 17, 333–351 (2016). https://doi.org/10.1038/nrg.2016.49
Logsdon, G.A., Vollger, M.R. & Eichler, E.E. Long-read human genome sequencing and its applications. Nat Rev Genet 21, 597–614 (2020). https://doi.org/10.1038/s41576-020-0236-x
https://journals.plos.org/plosone/article?id=10.1371/journal.pone.0163962
這個月的規劃貼在這篇文章中我們的基因體時代-AI, Data和生物資訊 Overview,也會持續調整!我們的基因體時代是我經營的部落格,如有對於生物資訊、檢驗醫學、資料視覺化、R語言有興趣的話,可以來交流交流!


 iThome鐵人賽
iThome鐵人賽