
上一篇我們的基因體時代-AI, Data和生物資訊 Day23- 基因註釋資料在Bioconductor中視覺化之呈現:Gviz我們示範如何將前幾天的知識整合一起,先使用AnnotationHub來取用人類基因組相關的資料包(org.Hs.eg.db, TxDb.Hsapiens.UCSC.hg38.knownGene, TxDb.Hsapiens.),再將這些資料轉換成Bioconductor中用來分析基因資訊的資料結構IRanges, GRanges,最後使用Gviz視覺化來比較舊版本和新版本中RhD基因的資料差異。
接著我們再往下如何用tidyverse的資料科學語法來分析前幾篇的基因資料,其實在前幾天的代碼中就使用蠻多tidyverse原生的library,但在處理IRanges和GRanges時卻還是非tidyverse的寫法,相對地不太順暢和直覺,也許不是每個人會使用R語言的人都聽過tidyverse,這邊先稍微介紹一下。
R語言在這10年多其實越來越流行,雖然這三年在深度學習風潮後比較退火外,在資料科學家間其實是蠻好用的一個語言,一方面跟他友善資料處理有關,R語言這十年來如此受歡迎,第一個是其原生是由統計學家們所開發的程式語言,所以很多統計工具都有相關的程式包,第二個則是語法上的進步和友善的社群,很多不錯的工具像是ggplot2, dplyr等等,算是讓人很容易就上手,且可以很functional programming來做分析,而統計學家Hadley Wickham便是這幾個重要library的開發者,其陸續形成了一個很棒的開發社群,同時還有商業化的公司Rstudio,提供不斷進步的R語言編輯器,其中在Hadley Wickham的領導下,陸陸續續把資料分析會用到的工具大抵上都開發完善如dplyr, readr, stringr, purrr, tidyr, tibble等,最後他把這些整合到一個包,叫做tidyverse,這樣一次就可以滿足百分之八十以上的資料分析需求,在這個開發體系中有幾個蠻好的原則,可參考這篇由Hadley Wickham撰寫的文章The tidy tools manifesto:
這個包的開發者Michael Lawrence其實就是當初開發IRange和GenomicRanges的人,所以可以理解他也會是把IRanges/GenomicRanges結構語法跟tidyverse整合的最佳人選,這個包建立的一些跟tidyverse一致的語法及跟基因資料處理相關的部分整合如下: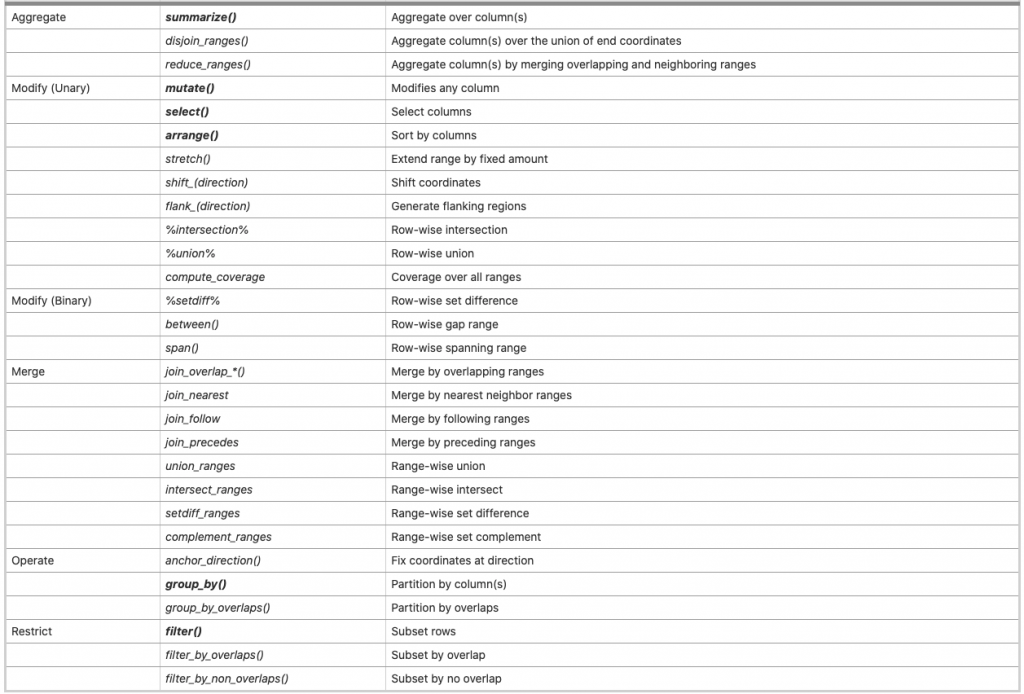
上面表格黑色粗體的部分就是沿襲自tidyverse中dplyr的語法:mutate, filter, summarize, groupy_by, select, arrange,其他則是跟IRange/GRange處理特異性相關的操作。
這邊承襲昨天的代碼,先取得多個跟輸血醫學相關的基因資訊:
library(org.Hs.eg.db)
library(TxDb.Hsapiens.UCSC.hg38.knownGene)
TxDb.hg38 <- TxDb.Hsapiens.UCSC.hg38.knownGene
gene.inform.df <- AnnotationDbi::select(org.Hs.eg.db,
keys = c("RHD", "VWF", "ADAMTS13"),
keytype = c("SYMBOL"),
columns = c("ENTREZID"))
hg38.cd.range.inform.df <- AnnotationDbi::select(TxDb.hg38,
keys = gene.inform.df$ENTREZID,
keytype = c("GENEID"),
columns = c("CDSCHROM", "CDSSTART","CDSID", "CDSEND", "CDSID","CDSSTRAND"))
相對於使用IRanges或是GRanges來建立物件,plyranges提供一個接近tidyverse語法的作法,使用as_ranges和as_granges函數,就能將data.frame的資料,轉換成IRanges和GRanges資料物件,但要遵守幾個原則,要轉換為iranges的data.frame,欄位至少要有start和end兩欄,而使用as_grangrs轉換為GRanges的物件,其data.frame至少要有三欄:start, end, seqnames,名字要一模一樣,就可以成功轉換,的確跟昨天和前天的語法比較一下,整個輕鬆簡單許多。
# 使用as_iranges
blood.irgs <- hg38.cd.range.inform.df %>%
dplyr::select(CDSSTART, CDSEND) %>%
dplyr::rename("start"=CDSSTART, 'end'=CDSEND) %>%
as_iranges()
# 使用as_granges
blood.grgs <- hg38.cd.range.inform.df %>%
dplyr::select(CDSCHROM, CDSSTART, CDSEND, GENEID, CDSSTRAND) %>%
dplyr::rename("start"=CDSSTART, 'end'=CDSEND, 'seqnames'=CDSCHROM) %>%
as_granges()
>blood.irgs
IRanges object with 139 ranges and 0 metadata columns:
start end width
<integer> <integer> <integer>
[1] 25272548 25272695 148
[2] 25284573 25284759 187
[3] 25290641 25290791 151
[4] 25300946 25301093 148
[5] 25301520 25301686 167
... ... ... ...
[135] 255794 256004 211
[136] 281379 281529 151
[137] 256126 256195 70
[138] 259472 259511 40
[139] 282207 282309 103
>blood.grgs
GRanges object with 139 ranges and 2 metadata columns:
seqnames ranges strand | GENEID CDSSTRAND
<Rle> <IRanges> <Rle> | <character> <character>
[1] chr1 25272548-25272695 * | 6007 +
[2] chr1 25284573-25284759 * | 6007 +
[3] chr1 25290641-25290791 * | 6007 +
[4] chr1 25300946-25301093 * | 6007 +
[5] chr1 25301520-25301686 * | 6007 +
... ... ... ... . ... ...
[135] chr9_KN196479v1_fix 255794-256004 * | 11093 +
[136] chr9_KN196479v1_fix 281379-281529 * | 11093 +
[137] chr9_KN196479v1_fix 256126-256195 * | 11093 +
[138] chr9_KN196479v1_fix 259472-259511 * | 11093 +
[139] chr9_KN196479v1_fix 282207-282309 * | 11093 +
-------
seqinfo: 4 sequences from an unspecified genome; no seqlengths
這邊我覺得超級方便,可以直接導入dplyr中filter的語法,針對任一個column的數值做篩選,可以是數值上的大小,或是字串上面的異同,比如我可以使用在IRanges的資料上,篩選出只有片段大於200的序列,同樣的,在比較資料複雜的GRanges物件,則可以做出如只要為在特定染色體上,或是後面meta data欄位的資訊,只是它的速度比想像中還慢,所以在較大的資料量下,可能要稍微等比較久一點。
> blood.irgs %>% filter(width > 200)
IRanges object with 17 ranges and 0 metadata columns:
start end width
<integer> <integer> <integer>
[1] 25328898 25329136 239
[2] 6110374 6110582 209
[3] 6075335 6075551 217
[4] 6056857 6057072 216
[5] 6052543 6052783 241
... ... ... ...
[13] 133429700 133429910 211
[14] 280509 280713 205
[15] 281379 281697 319
[16] 285068 285274 207
[17] 255794 256004 211
> blood.grgs %>% filter(GENEID == '6007')
GRanges object with 15 ranges and 2 metadata columns:
seqnames ranges strand | GENEID CDSSTRAND
<Rle> <IRanges> <Rle> | <character> <character>
[1] chr1 25272548-25272695 * | 6007 +
[2] chr1 25284573-25284759 * | 6007 +
[3] chr1 25290641-25290791 * | 6007 +
[4] chr1 25300946-25301093 * | 6007 +
[5] chr1 25301520-25301686 * | 6007 +
... ... ... ... . ... ...
[11] chr1 25317000-25317052 * | 6007 +
[12] chr1 25328898-25328924 * | 6007 +
[13] chr1 25321889-25321962 * | 6007 +
[14] chr1 25328898-25329136 * | 6007 +
[15] chr1 25328898-25329015 * | 6007 +
-------
seqinfo: 4 sequences from an unspecified genome; no seqlengths
可以直接使用mutate來修改物件中的欄位,而其他欄位會同時調整,比如你修改這個基因區段的長度,那麼你則可已修改width,則start或end的位置會調整,可以使用anchor_start, anchor_end, anchor_centre, anchor_5p, anchor_3p來指定這樣的調整,要以哪邊為基準。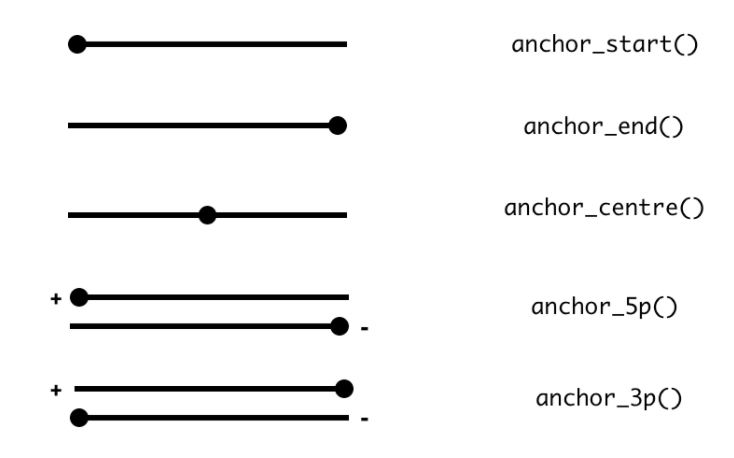
可以看一下面的示範:
> blood.irgs %>% mutate(width=10)
IRanges object with 139 ranges and 0 metadata columns:
start end width
<integer> <integer> <integer>
[1] 25272548 25272557 10
[2] 25284573 25284582 10
[3] 25290641 25290650 10
[4] 25300946 25300955 10
[5] 25301520 25301529 10
... ... ... ...
[135] 255794 255803 10
[136] 281379 281388 10
[137] 256126 256135 10
[138] 259472 259481 10
[139] 282207 282216 10
> blood.irgs %>% anchor_end() %>% mutate(width=10)
IRanges object with 139 ranges and 0 metadata columns:
start end width
<integer> <integer> <integer>
[1] 25272686 25272695 10
[2] 25284750 25284759 10
[3] 25290782 25290791 10
[4] 25301084 25301093 10
[5] 25301677 25301686 10
... ... ... ...
[135] 255995 256004 10
[136] 281520 281529 10
[137] 256186 256195 10
[138] 259502 259511 10
[139] 282300 282309 10
> blood.irgs %>% anchor_start() %>% mutate(width=10)
IRanges object with 139 ranges and 0 metadata columns:
start end width
<integer> <integer> <integer>
[1] 25272548 25272557 10
[2] 25284573 25284582 10
[3] 25290641 25290650 10
[4] 25300946 25300955 10
[5] 25301520 25301529 10
... ... ... ...
[135] 255794 255803 10
[136] 281379 281388 10
[137] 256126 256135 10
[138] 259472 259481 10
[139] 282207 282216 10
> blood.irgs %>% anchor_center() %>% mutate(width=10)
IRanges object with 139 ranges and 0 metadata columns:
start end width
<integer> <integer> <integer>
[1] 25272617 25272626 10
[2] 25284661 25284670 10
[3] 25290711 25290720 10
[4] 25301015 25301024 10
[5] 25301598 25301607 10
... ... ... ...
[135] 255894 255903 10
[136] 281449 281458 10
[137] 256156 256165 10
[138] 259487 259496 10
[139] 282253 282262 10
除了基礎的建立物件和篩選,其實還能做超多方便的運算這部分真的蠻不錯的!
Lee, S., Cook, D. & Lawrence, M. plyranges: a grammar of genomic data transformation. Genome Biol 20, 4 (2019). https://doi.org/10.1186/s13059-018-1597-8
Chapter 16. Visualizing Genomic Data Using Gviz and Bioconductor. Statistical Genomics. 2016
Ou J, Zhu LJ (2019). “trackViewer: a Bioconductor package for interactive and integrative visualization of multi-omics data.” Nature Methods, 16, 453–454. doi: 10.1038/s41592-019-0430-y, https://doi.org/10.1038/s41592-019-0430-y.
這個月的規劃貼在這篇文章中我們的基因體時代-AI, Data和生物資訊 Overview,也會持續調整!我們的基因體時代是我經營的部落格,如有對於生物資訊、檢驗醫學、資料視覺化、R語言有興趣的話,可以來交流交流!
